Understanding how microbial genomes are organized in three-dimensional space is becoming a key part of microbiome research. Traditional sequencing tells us what genes are present, but it often loses the physical context of how those genes are arranged inside cells. Meta-3C (Chromosome Conformation Capture) was developed to bridge this gap. By capturing the physical contacts between DNA fragments, this method allows scientists to explore not only which genomes exist in a mixed community, but also how they are structured and connected.
In this article, we introduce the principles of meta-3C, how it differs from Hi-C and other 3C-based methods, its experimental workflow, and the advantages it offers for microbiome research. Our aim is to provide a clear, research-focused guide for scientists interested in applying spatial genomics tools to complex microbial communities.
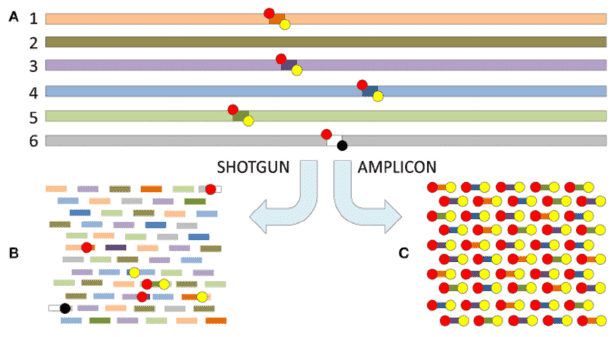
 meta3C experiment on a controlled mix of bacterial species. (Marbouty, Martial, et al. elife 2014)
meta3C experiment on a controlled mix of bacterial species. (Marbouty, Martial, et al. elife 2014)
How meta-3C Differs from Hi-C
Chromosome Conformation Capture (3C) technologies have become essential tools for exploring the three-dimensional organization of genomes. Among them, Hi-C is widely used to map DNA-DNA contacts across entire genomes. It has been applied successfully in single organisms, providing high-resolution maps of chromatin interactions. However, applying Hi-C directly to microbial communities presents challenges. In mixed samples, DNA from different species is present at once, and standard Hi-C pipelines struggle to assign sequencing reads to the correct genome.
This is where meta-3C provides a unique advantage. Developed as an adaptation of Hi-C for complex microbial ecosystems, meta-3C can capture spatial interactions between DNA fragments in mixed samples and use this information to group them into species-specific bins. In other words, while Hi-C works best when the genome of interest is already known, meta-3C enables genome assembly and structural analysis without prior reference sequences.
Researchers have reported that meta-3C is especially effective in:
- Resolving microbial genomes from environmental samples where many organisms remain uncultured.
- Linking plasmids and mobile genetic elements to their host chromosomes, a task that conventional sequencing often leaves unresolved.
- Improving metagenomic binning by reducing contamination and enhancing genome completeness.
By design, meta-3C does not replace Hi-C but rather extends its scope into microbiome research. For scientists working with soil, marine, or gut microbial communities, it offers a practical way to connect DNA sequence data with the physical structure of genomes in their native environment.
Applying meta-3C in Mixed Microbial Communities
In complex microbial communities—such as soil, marine environments, or host-associated microbiomes—traditional metagenomic approaches often fall short at reconstructing complete genomes from mixed DNA. That's because these methods disrupt spatial context and make it challenging to assign DNA fragments to their source organisms reliably.
Enter meta-3C, a powerful adaptation of chromosome conformation capture tailored for metagenomics. Developed by Marbouty and colleagues, meta-3C uses proximity-ligation signals between DNA fragments to group them by physical co-association. By applying meta-3C to controlled mixtures of bacteria and yeast, the team was able to assemble, scaffold, and infer three-dimensional genome architecture—even for species without prior reference genomes.
Another independent study tested the meta-3C approach using mock microbial communities. Liu and Darling reproduced the protocol, suggested simple optimizations, and compared meta-3C to standard Hi-C. They found that meta-3C delivered high-quality data with a more straightforward workflow, making it an efficient choice for spatially resolved metagenomic analysis.
A practical example of its impact comes from the MetaTOR pipeline. Applied to 20 meta-3C libraries derived from mouse gut microbiota, MetaTOR recovered 82 high-quality metagenome assembled genomes (MAGs)—a significant improvement compared to just 9 MAGs from a single library. This result illustrates how combining meta-3C with optimized binning can dramatically enhance genome recovery from complex samples.
 MetaTOR pipeline. (Baudry, Lyam, et al., Frontiers in genetics 2019)
MetaTOR pipeline. (Baudry, Lyam, et al., Frontiers in genetics 2019)
Why This Matters for Researchers
- De novo genome reconstruction: meta-3C enables assembly and scaffolding without requiring prior reference sequences.
- Improved binning accuracy: Spatial linkage helps distinguish DNA fragments from different organisms, reducing misassignment.
- Structural insights: Beyond sequence identity, meta-3C reveals how chromosomes and mobile elements are spatially organized within cells.
Core Principles and Workflow of meta-3C
Understanding how meta-3C works at a technical level is key for laboratory scientists looking to adopt this method. At its core, meta-3C adapts standard 3C-based workflows to preserve—and then exploit—the spatial proximity information of DNA fragments in mixed microbial samples.
 3C and chromatin interaction methods. (Liu, Michael, and Aaron Darling., F1000Research 2015)
3C and chromatin interaction methods. (Liu, Michael, and Aaron Darling., F1000Research 2015)
Key Experimental Steps in meta-3C
1. Crosslinking (Fixation)
Samples are treated with formaldehyde (typically 1–3%) to freeze DNA–DNA and DNA–protein interactions in place. This step preserves the three-dimensional arrangement of genomes within each microbial cell, which is then exploited downstream.
2. Restriction Digestion
Crosslinked chromatin is cut with a restriction enzyme (e.g., HindIII or EcoRI) to fragment DNA at specific recognition site. In the context of meta-3C, these fragments retain spatial linkage information within microbial cells.
3. Chromatin Interaction
Under dilute conditions, fragment ends that were crosslinked in close proximity ligate to each other, forming novel DNA junctions that capture spatial contacts.
4. Reverse Crosslinking & DNA Purification
Crosslinks are reversed (typically via heat or proteinase treatment), and the DNA is purified. This yields chimeric fragments—each containing sequences that were physically adjacent in the original sample.
5. Sequencing Library Preparation & Sequencing
Purified chimeric DNA is processed into a sequencing-ready library. Sequencing reads from these junctions are key in reconstructing spatial relationships using computational pipeline.
6. Data Analysis & Genome Reconstruction
Computational tools—like MetaTOR—use the spatial linkage information to cluster sequences by species or strain, improve scaffolding, and reconstruct MAGs by leveraging three-dimensional contact data.
Why This Workflow Matters
- Preserves Spatial Context: Unlike traditional metagenomic workflows that disrupt linkage during sample preparation, meta-3C retains physical DNA proximity, enabling new ways to group and assemble sequences.
- Enables De Novo Reconstruction: The spatial signal allows for genome assembly without relying on reference genomes—especially valuable in studies of uncultured or novel microbes.
- Improves Binning and Assembly Quality: Meta-3C delivers more accurate binning and scaffolding by leveraging physical DNA interactions, significantly reducing contamination and chimerism.
Summary Table
| Step | Description |
|---|---|
| Crosslinking | Preserve spatial DNA interactions using formaldehyde |
| Digestion | Fragment DNA while retaining spatial linkage |
| chromatin interaction | Capture interacting fragments by ligating together |
| Reverse Crosslinking | Free DNA for sequencing after unlinking |
| Sequencing | Sequence junction reads to capture spatial info |
| Computational Analysis | Cluster and assemble genomes using spatial links |
Advantages of meta-3C in Microbiome Research
For researchers working with complex microbial communities, meta-3C offers several compelling advantages over traditional metagenomic methods:
1. De Novo Assembly Without Reference Genomes
A single meta-3C experiment can be used to assemble, scaffold, and reveal the three-dimensional organization of genomes—without relying on preexisting reference sequences. This allows the reconstruction of unknown or uncultured microbial genomes from environmental or host-associated samples.
2. Preservation of Spatial Context
Unlike conventional metagenomic workflows that break down DNA into fragments and lose physical linkage, meta-3C preserves spatial proximity information, enabling researchers to exploit physical DNA interactions to group sequences into biologically meaningful assemblies.
3. Higher-Quality Genome Recovery
When applied with pipelines like MetaTOR, meta-3C significantly improves metagenome-assembled genome (MAG) recovery. In one study of mouse gut microbiota, MetaTOR identified 82 high-quality MAGs from 20 meta-3C libraries, compared to just 9 MAGs from a single library—demonstrating a threefold increase in performance over traditional binning tools.
4. Linking Mobile Genetic Elements to Host Genomes
meta-3C can associate plasmids, phages, and other mobile genetic elements with their host chromosomes—an analysis difficult or impossible with standard short-read metagenomics.
Conclusion & Future Directions
As we've seen, meta-3C stands out as a remarkable tool for exploring the spatial organization of microbial genomes in complex samples—enabling de novo assembly, preserving physical context, and improving binning quality. But what lies ahead for this emerging technology?
1. Integrating with Long-Read 3D Techniques (e.g., Pore-C)
Recent advancements in chromatin conformation capture—such as Pore-C, which leverages long-read nanopore sequencing—offer higher-order interaction detection and simplified workflows. Pore-C allows the capture of multi-fragment concatemers without amplification and even retains methylation information, illuminating epigenetic landscapes alongside 3D structure.
Adapting Pore-C principles into meta-3C protocols could enhance resolution, enable detection of long-range structural connections, and integrate spatial arrangement with epigenetic context in microbial genomes.
2. Standardizing High-Throughput and Multi-Omic Meta-3C Pipelines
While meta-3C shows great promise in manual or small-scale workflows, future efforts should focus on developing robust, scalable pipelines. Combining automated sample processing, improved noise-reduction algorithms, and multi-omic integration—such as linking spatial data with transcriptomics or metabolomics—could unlock deeper insights into microbial ecology and function.
3. Deepening Understanding of Microbial Chromosome Architecture
The original meta-3C study using mixed bacterial/yeast communities revealed significant diversity in microbial chromosome organization. Future research could expand this exploration across environments—soil, marine ecosystems, human microbiomes—helping us understand how spatial genome architecture affects microbial behavior, symbiosis, and evolution.
4. Bridging Structural Genomics and Functional Dynamics
Enhanced meta-3C applications could link 3D genomic structure with functional states—such as replication timing, gene expression, or stress response. Insights from higher-resolution techniques (like Hi-C in eukaryotes) have reshaped our understanding of genome regulation. Applying analogous strategies to microbial communities may uncover connections between spatial genome folding and ecological adaptation.
Concluding Thoughts
meta-3C represents a significant leap in metagenomic methods—merging spatial genome architecture with community-scale sequencing. Looking ahead, integrating emerging technologies like Pore-C, automating workflows, expanding environmental applications, and connecting structure with function will solidify meta-3C's status as a powerful, future-proof tool for microbiome research.
How CD Genomics Supports meta-3C Research
CD Genomics offers a streamlined Meta Hi-C / 3C service to empower researchers in studying the spatial organization of microbial genomes:
- Improved genome assembly: By clustering DNA sequences based on physical proximity, this technology enhances metagenome assembly at the species level—especially beneficial in complex environmental samples.
- Linking mobile elements to hosts: The service excels at associating plasmids, phages, and resistance genes with their microbial hosts—a key factor for tracking horizontal gene transfer.
- Simplified sample preparation: For each sample, CD Genomics constructs three 3C libraries using four-base cutter, avoiding biotin labeling. This simplifies the workflow while improving GC content coverage across diverse microbes.
(Research use only—this service isn't for clinical or personal diagnostics.)
References
- Marbouty, Martial, et al. "Metagenomic chromosome conformation capture (meta3C) unveils the diversity of chromosome organization in microorganisms." elife 3 (2014): e03318.
- Liu, Michael, and Aaron Darling. "Metagenomic Chromosome Conformation Capture (3C): techniques, applications, and challenges." F1000Research 4 (2015): 1377.
- Baudry, Lyam, et al. "MetaTOR: a computational pipeline to recover high-quality metagenomic bins from mammalian gut proximity-ligation (meta3C) libraries." Frontiers in genetics 10 (2019): 753.
- Marbouty, Martial, and Romain Koszul. "Metagenome analysis exploiting high-throughput chromosome conformation capture (3C) data." Trends in Genetics 31.12 (2015): 673-682.
- DeMaere, Matthew Z., and Aaron E. Darling. "Sim3C: simulation of Hi-C and Meta3C proximity ligation sequencing technologies." GigaScience 7.2 (2018): gix103.