
 Sample Submission Guidelines
Sample Submission Guidelines
In gene medicine, variant analysis helps doctors turn DNA data into useful health insights. The process starts by filtering out common gene changes using databases like gnomAD. Then, tools like CADD and REVEL predict if the remaining variants might cause problems. Other tools, such as SIFT and PolyPhen-2, check for protein damage, while SpliceAI finds splicing errors. Medical guidelines (ACMG) classify variants as harmful, harmless, or uncertain. This classification impacts patient care. For example, it helps. The biggest challenge is understanding unclear variants (VUS), which often need family health history or more lab tests to determine their real impact.
This paper looks at variant analysis methods, focusing on how genomic databases prioritize variants and how guidelines help apply them in precision medicine.
Overview of Variant Analysis Workflow
Variant analysis is now a key part of precision medicine. It helps find and understand disease-related gene changes among thousands of variants. The process turns raw DNA data into useful medical insights through several steps. First, advanced sequencing captures detailed genetic information, creating large datasets. Then, special tools analyze this data to spot differences from a standard reference genome. The variant analysis workflow typically has several main stages:
- Data generation: Obtaining high-quality sequence reads through various sequencing approaches
- Alignment: Mapping reads to a reference genome
- Variant calling: Identifying differences between the subject’s genome and the reference
- Quality control: Filtering out technical artifacts and low-confidence calls
- Annotation: Adding functional and contextual information to each variant
- Prioritization: Ranking variants based on multiple criteria
- Interpretation: Assessing the clinical significance of prioritized variants
This step-by-step approach helps analysts focus on the most promising candidates among the many variants in each person’s genome, creating a smaller, more manageable group for detailed study.
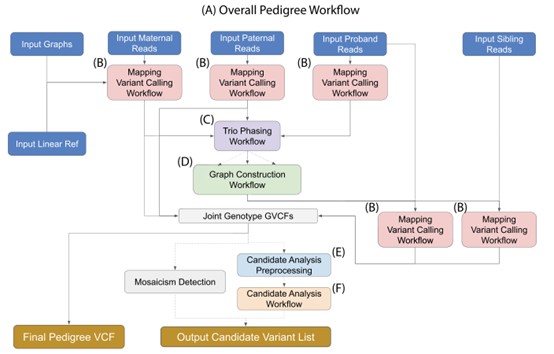
Figure 1. Single-sample alignment and variant calling workflow. ( Markello, 2022)
Filtering Based on Frequency and Function
Population Allele Frequency Filtering
The first key step is filtering out common gene variants. Researchers use large population databases like gnomAD and ExAC, which contain genetic data from hundreds of thousands of people. These databases help determine how often variants appear in the general population.
Key filtering rules:
- For rare diseases: Exclude variants found in more than 1% (or sometimes 0.1%) of people
- For common diseases: May keep more frequent variants
- Must consider ethnicity, as some variants are more common in certain groups
These databases (like gnomAD with 125,000+ exomes and 15,000+ genomes) help scientists tell the difference between harmless common variants and rare ones that might cause disease. The diverse data helps ensure accurate results across different populations.
Functional Scoring Tools
Multiple computational tools assess the potential functional importance of variants:
- CADD: Integrates multiple annotations into a single score, with higher scores suggesting greater deleteriousness
- REVEL: Specifically designed for rare missense variants, combining multiple prediction algorithms
- MetaSVM: Employs machine learning approaches to integrate various prediction algorithms
- FATHMM: Predicts the functional consequences of both coding and non-coding variants
These tools provide quantitative metrics for variant prioritization, helping researchers focus on variants most likely to have biological significance. For instance, a CADD score above 20 places a variant in the top 1% of predicted deleterious variants in the human genome, making it a strong candidate for further investigation.
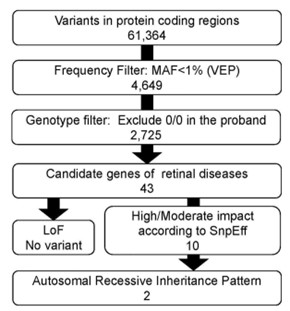
Figure 2. The overview of variant filtering steps. ( Saito, 2021)
Variant Analysis for Predicting Functional Consequences
Amino Acid Change Prediction Tools
For coding region variants, assessing their impact on protein function is essential:
- SIFT: Evaluates whether an amino acid substitution affects protein function based on sequence homology and physical properties
- PolyPhen-2: Combines structural and comparative genomics approaches to predict variant impact
- MutationTaster: Evaluates the effect of DNA sequence alterations on protein function, considering evolutionary conservation and protein domain information.
- PROVEAN: Predicts whether an amino acid substitution or indel has an impact on the biological function of a protein.
These tools calculate scores based on different algorithms and training datasets, each with unique strengths. For example, SIFT primarily utilizes sequence conservation, while PolyPhen-2 incorporates protein structure data when available. Multiple prediction tools often provide more robust assessments than any single algorithm.
Splice Site Prediction Algorithms
Splicing variants can lead to abnormal RNA processing, even if they don’t directly alter coding sequences:
- SpliceAI: A deep learning model that predicts splice junctions from an arbitrary pre-mRNA transcript sequence
- MaxEntScan: Evaluates how variants change the strength of splice sites
- Human Splicing Finder: Predicts the effects of mutations on splicing signals
- NNSplice: Uses neural networks to predict splice sites
These prediction tools help find important variants that might be missed by regular analysis, especially those affecting gene activity rather than protein structure. Splicing variants can cause exon skipping, intron retention, or trigger hidden splice sites. These changes can lead to major problems with how genes work, even though they occur outside the main coding regions.
Variant Analysis for Clinical Interpretation Frameworks
ACMG Guidelines for Variant Classification
The American College of Medical Genetics and Genomicshas established a standardized framework that classifies variants into five categories:
- Pathogenic
- Likely pathogenic
- Variant of uncertain significance
- Likely benign
- Benign
This classification is based on multiple lines of evidence, including:
- Population data (prevalence and case-control studies)
- Computational predictions (in silico analysis)
- Functional studies
- Segregation data
- De novo occurrence
- Allelic data
- Other database entries
- Literature reports
The ACMG guidelines provide specific criteria (PS1-4, PM1-6, PP1-5, BA1, BS1-4, BP1-7) that can be combined to reach a final classification. This systematic approach standardizes interpretation across laboratories and improves consistency in clinical reporting.
Relevance in Diagnostics and Pharmacogenomics
Variant interpretation has two primary applications in clinical settings:
- Diagnostic applications: Identifying variants associated with specific disease phenotypes to establish molecular diagnoses
- Pharmacogenomic applications: Recognizing variants affecting drug metabolism, transport, or targets to guide individualized treatment plans
These applications require rigorous evidence standards and careful clinical judgment to ensure genetic information appropriately guides medical decisions. The threshold of evidence may differ between these applications; for instance, pharmacogenomic variants often have more direct functional evidence but may require specific considerations about drug-gene interactions and clinical actionability.
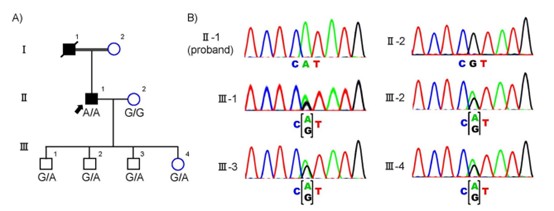
Figure 3. Genetic analysis of the consanguineous family with retinitis pigmentosa. ( Saito, 2021)
Challenges in Clinical Reporting
Variant of Uncertain Significance (VUS) Dilemmas
A significant challenge is managing the large number of variants of uncertain significance:
- VUS cannot be used for clinical decision-making, but may contain important information
- VUS may be reclassified as knowledge accumulates
- Communicating VUS information to patients presents complex ethical considerations
Reports suggest that approximately 40-60% of unique variants identified in clinical testing are classified as VUS. This creates substantial challenges for genetic counseling and patient education. Laboratories must establish clear protocols for periodic reassessment of VUS and notification of healthcare providers when reclassifications occur.
Need for Functional Validation and Segregation Analysis
Key strategies to improve the reliability of variant interpretation include:
- Laboratory functional validation studies confirming the impact of variants on protein function
- Family segregation analysis observing whether variants co-inherit with disease phenotypes
- Multi-omics integration approaches, such as combining transcriptomic or proteomic data
- International collaboration and data sharing to accumulate additional evidence
Functional studies are particularly valuable for resolving VUS classifications. These may include assays measuring enzyme activity, protein localization, binding properties, or pathway activation. For example, missense variants in tumor suppressor genes might be evaluated through colony formation assays, while ion channel variants could be assessed using electrophysiology studies.
Segregation analysis provides powerful evidence when multiple affected and unaffected family members are available for testing. Finding that a variant perfectly segregates with disease across multiple generations strengthens the case for pathogenicity, particularly for highly penetrant conditions.
Conclusion
Variant analysis changes raw genetic data into useful medical insights. These insights help inform patient care. Healthcare professionals filter, predict, and interpret DNA variations. This way, they can uncover valuable health insights from complex genetic test results. This important field connects lab results to clinical use. It helps doctors find disease risks, pick the right treatments, and provide personalized care based on a person’s genes.
The process begins when genetic testing identifies thousands of variations in a person’s DNA. Medical teams then systematically analyze these variations to determine which might affect health. They filter out common, harmless variations. They focus on gene changes linked to specific conditions. They predict how each change may affect protein function. They also interpret findings based on the patient’s health history and symptoms.
This field is experiencing remarkable growth and advancement due to several technological breakthroughs:
- Next-generation DNA sequencing has dramatically reduced the cost and increased the speed of genetic testing, making comprehensive genetic analysis more accessible to patients
- Sophisticated computational tools can now process vast amounts of genetic data quickly, identifying patterns and correlations that would be impossible to detect manually
- Expanding genetic databases collect information from diverse populations worldwide, improving our understanding of genetic variations across different ethnic backgrounds
Despite these advances, significant challenges continue to shape the landscape of variant analysis:
- Dealing with unclear results (VUS)
- Understanding why symptoms vary between people
- Explaining why some genetic changes don’t always cause disease
These challenges show that genomic medicine mixes science with clinical judgment. Good care requires teamwork. Genetic experts, lab scientists, and doctors must work together. Ongoing learning and strong quality standards are also important.
As research grows, variant analysis will become even more important for personalized medicine. It will help doctors and patients make better health choices based on each person’s unique genetic makeup. Turning DNA data into medical advice is tough. Still, each new discovery helps us move toward personalized healthcare.
References:
- Markello, C., Huang, C., et,al. (2022). A complete pedigree-based graph workflow for rare candidate variant analysis. Genome research, 32(5), 893–903. https://doi.org/10.1101/gr.276387.121
- Saito, K., Gotoh, N., et, al. (2021). A case of retinitis pigmentosa homozygous for a rare CNGA1 causal variant. Scientific reports, 11(1), 4681. https://doi.org/10.1038/s41598-021-84098-9