
 Sample Submission Guidelines
Sample Submission Guidelines
In the vast human genome, variations are everywhere. Each person’s genome carries millions of small differences in sites. These genomic variations not only shape physiological differences and disease susceptibility between individuals, but also profoundly affect the evolution of mankind. With the rapid development of genomics and high-throughput sequencing technology, scientists are able to more systematically identify, classify and analyze these variations, revealing their important role in health, disease and even human history.
This article will systematically introduce the main types of genomic variations and their detection methods, deeply explore the potential effects of different variations on gene function and protein structure, and explain the biological significance of genomic variations in genetic diseases and human evolution.
Classification of Genome Variants
In genomic research, identifying and analyzing genomic variations represents a critical field of study. Methods for detecting and classifying various genome alterations—such as single nucleotide polymorphisms (SNPs), insertions and deletions (indels), variations in copy number (CNVs), and structural variants (SVs)—form the foundation of this discipline. The significance of these genomic differences extends across multiple domains including studies of hereditary disorders, evolution-focused investigations, and the emerging field of individualized therapeutic approaches.
Single Nucleotide Polymorphism
SNPs refer to changes in a single base and are the most common type of variation in the genome. They can affect protein coding, transcriptional regulation, and the function of non-coding RNA. SNPs are widely used in disease susceptibility research, pharmacogenomics, and human evolution research .
Indels
Indels refer to the insertion or deletion of short DNA fragments (usually less than 50 bp) in the genome. Such variations may affect gene function, such as changing splicing sites or causing frameshift mutations.
Copy number variation
CNVs refer to changes in the number of copies of a gene or DNA segment, including duplications and deletions. CNVs have a significant impact on the susceptibility to genetic diseases and are also of great significance in population genetics.
Structural variation
SVs include insertions, deletions, inversions, translocations, and duplications, and usually involve larger DNA fragments (>1kb). SVs have a profound impact on genome stability and function and are closely related to the occurrence of many complex diseases.
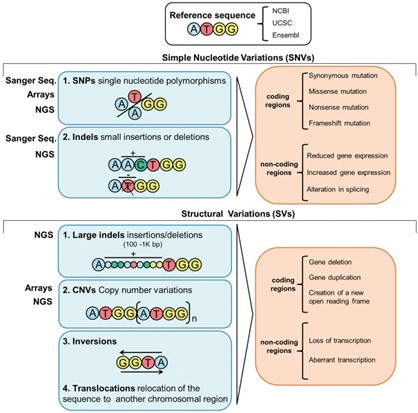
Figure 1. Overview of the types of variants in the genome.( Manzoni, C., et al . 2018 )
Functional Impacts on Genes and Proteins
The effects of genomic variation on gene and protein function are mainly reflected in synonymous mutations, nonsynonymous mutations, nonsense mutations, and frameshift mutations, etc. These mutation types have different effects in changing gene expression, protein structure, and function.
Synonymous vs. nonsynonymous mutations
Synonymous mutation: A synonymous mutation refers to a change in the DNA sequence, but the encoded amino acid has not changed. Such mutations are generally considered “silent” because they do not directly affect the amino acid sequence of the protein. However, studies in recent years have shown that synonymous mutations may indirectly affect protein expression and function by affecting transcription factor binding sites, mRNA stability, splicing, and RNA folding. For example, some synonymous mutations may change mRNA levels, thereby indirectly affecting protein expression. In addition, the role of synonymous mutations in diseases has gradually been valued, and they may be associated with susceptibility to complex diseases .
Non-synonymous mutations: Non-synonymous mutations change the amino acid sequence of a protein and may result in loss or alteration of protein function. Such mutations are generally considered to have a greater impact on individuals because they directly change the structure and function of the protein. Non-synonymous mutations include missense mutations and in-frame insertions/deletions, which may cause severe changes in protein function, such as altering protein activity, stability, or folding.
Nonsense and frameshiftmutations
Nonsense mutations: Nonsense mutations lead to premature termination of translation by introducing premature stop codons (such as TAA, TGA, or TAG), thereby generating truncated, nonfunctional proteins. Such mutations often lead to severe phenotypic consequences because they disrupt the integrity of the protein .
Frameshift mutations: Frameshift mutations are caused by the insertion or deletion of one or more nucleotides in the DNA sequence, changing the reading frame and thus affecting the translation of the entire gene. This mutation may result in a completely different protein sequence, usually rendering the protein nonfunctional or producing a new, dysfunctional protein. Frameshift mutations are considered to be more serious types of mutations because of their wide-ranging effects on gene expression and protein function.
High-throughput functional screening technologies such as CRISPR-Cas9 provide powerful tools for studying the pathogenic effects of nonsense mutations and frameshift mutations. Through CRISPR-Cas9 technology, these mutations can be accurately simulated in cells or animal models to observe their effects on cell function and phenotype. At the same time, this technology can also be used for gene editing to try to repair mutations, providing potential methods for treating related diseases.
Genome Variants in Human Disease
Genomic variation plays a key role in regulating human health and disease. Based on how variation affects disease mechanisms, it can generally be divided into two categories: monogenic mutations and polygenic risk loci. These two types of variation differ significantly in genetic mechanisms, disease characteristics, and clinical applications.
Monogenic mutations
Single gene mutation refers to a genetic disease caused by a pathological variation in a single gene. Such diseases often manifest as rare but severe clinical phenotypes with a clear inheritance pattern. Single gene mutations are usually caused by the replacement, deletion or insertion of a single base pair, which in turn leads to loss of function, structural abnormalities or disordered expression of the encoded protein. Depending on the way the mutation affects the trait, single gene diseases can be divided into dominant inheritance (such as Huntington’s disease) or recessive inheritance (such as cystic fibrosis) and follow Mendel’s laws of inheritance.
Although the overall incidence of monogenic diseases is low, they have become an important subject of genetic research due to their high pathogenicity and relatively clear molecular mechanisms. With the development of high-throughput sequencing technologies, such as whole exome sequencing (WES) and whole genome sequencing (WGS), the detection of single gene mutations has become more accurate and efficient, significantly improving the possibility of early clinical diagnosis and intervention. In addition, emerging therapies such as gene therapy and targeted therapy have shown good prospects in some monogenic diseases, such as directly repairing pathogenic mutations through CRISPR-Cas9 technology, or restoring abnormal protein function using small molecule drugs.
Polygenic risk loci
Unlike single-gene diseases, polygenic risk loci involve the joint action of multiple genes. Each locus contributes less to disease risk, but their cumulative effect can significantly affect an individual’s susceptibility to disease. This type of genetic variation is widely present in common complex diseases, such as cardiovascular disease, diabetes, Alzheimer’s disease, and various cancers. Polygenic risk loci are mostly located in the non-coding regions of the genome, and they indirectly affect the occurrence and development of diseases mainly by regulating cell or tissue-specific gene expression.
The risk assessment of polygenic diseases relies on the integration of information from multiple variant sites. Currently, the polygenic risk score (PRS) has become a research focus. By quantifying the cumulative contribution of multiple single nucleotide polymorphisms to disease risk, individuals with higher genetic risks can be identified at the population level. This method is mainly based on data from large-scale genome-wide association studies (GWAS) and has been initially applied to prediction and early intervention guidance in certain disease areas.
Evolutionary Role of Genome Variation
The role of genomic variation in evolution is a complex and diverse process, in which positive selection signals and founder effects are two key factors.
Positive selection signals
Positive selection refers to natural selection promoting the spread and fixation of beneficial mutations, thereby increasing the frequency of these beneficial genes in the population. This selection mechanism has played an important role in human evolution. For example, genes related to brain function, language ability, dietary adaptability, and pathogen resistance all show strong positive selection signals .
Lactose tolerance is a typical example of the spread of genomic variation through positive selection in humans. Under normal circumstances, lactase activity decreases with age, leading to lactose intolerance, but in some populations, genetic variation due to the continued expression of lactase is retained and amplified through positive selection of animal husbandry culture. Studies have shown that the main reason for the difference in lactose tolerance between regions and populations is a point mutation C/T−13910 at the rs4988235 site of the MCM6 gene. Although MCM6 does not directly synthesize lactase, it can directly affect the expression of the nearby lactase synthesis gene LCT as a regulatory element with enhancer-like functions. The mutation first appeared in the Central European steppe between 7500 and 5000 years ago. It then spread rapidly to other parts of Europe. We can find strong selection characteristics around the LCT and MCM6 genes in lactose-tolerant people.
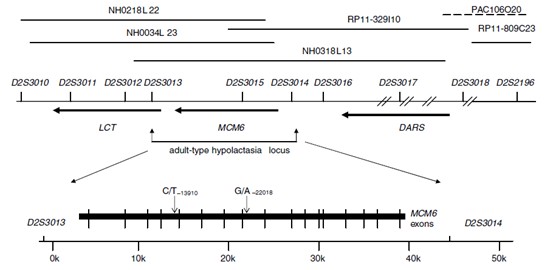
Figure 2. Genetic localization of lactose intolerance loci.(Enattah et al. 2002)
Founder effects
The founding effect refers to the phenomenon that when a new population is separated from a large population by a small number of individuals, the variability of its genome is significantly reduced. This effect can cause the gene frequencies of the new population to be significantly different from those of the original population, which may trigger significant changes in genetic characteristics. For example, the high incidence of certain genetic diseases in a specific population may be the result of the founding effect . The founding effect not only affects genetic diversity, but also may promote species differentiation and the formation of new species. For example, the African population migration event is considered to be the original founding effect, which has led to a significant reduction in genetic diversity in the modern human genome. In addition, the founding effect may also lead to rapid changes in the frequencies of certain genes, thereby affecting the genetic composition of the population.
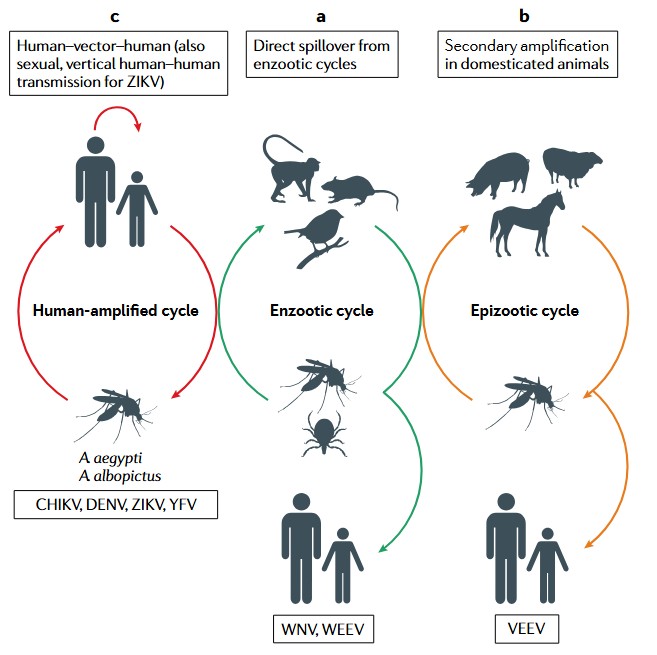
Figure 3. Arbovirus transmission cycles and emergence.(Weaver et al. 2021)
Challenges and Prospects in Genome Variation
Although significant progress has been made in the study of genomic variation, it still faces many challenges and also contains broad prospects for future development.
The interpretation of mosaic variants is a major challenge currently faced. Mosaic variants refer to the presence of different genomic variants in different cells of the same individual, which makes the detection and interpretation of variants extremely complicated. Since the variation in different tissues and cells may be different, traditional detection methods are difficult to accurately capture these differences. For example, in tumor tissue, there may be many different cell subpopulations, each with its own unique genomic variation. How to accurately identify and interpret these mosaic variants is crucial for the diagnosis and treatment of tumors.
The integration of multi-omics data is also an urgent problem to be solved. With the development of technology, we can obtain multi-omics data such as genome, transcriptome, and proteome. However, how to effectively integrate and analyze these data at different levels is a challenge currently faced. There are complex relationships between different omics data, and the data of a single omics often cannot fully reveal biological processes and disease mechanisms. For example, the expression level of a gene is not only affected by genomic variation, but also closely related to factors such as transcriptional regulation and protein modification. Therefore, it is necessary to develop more advanced algorithms and tools to achieve deep integration of multi-omics data.
For more related content, check out the following articles:
References:
- Manzoni, C., et al. (2018). Genome, transcriptome and proteome: the rise of omics data and their integration in biomedical sciences. Briefings in bioinformatics, 19(2), 286–302. https://doi.org/10.1093/bib/bbw114
- Sauna, Z. E., & Kimchi-Sarfaty, C. (2011). Understanding the contribution of synonymous mutations to human disease. Nature reviews. Genetics, 12(10), 683–691. https://doi.org/10.1038/nrg3051
- Enattah, N. S., et al. (2002). Identification of a variant associated with adult-type hypolactasia. Nature genetics, 30(2), 233–237. https://doi.org/10.1038/ng826