
 Sample Submission Guidelines
Sample Submission Guidelines
Variant Calling Format (VCF) is a standardized text file format widely used in bioinformatics and genomics to store and exchange genetic variation information. Researchers and clinicians depend on this standardized approach when analyzing differences in DNA sequences across populations or individuals. The importance of this format extends across multiple domains of genetic research and personalized medicine.This article will elaborate on VCF’s structure, tools, clinical applications, and file processing.
Introduction to Variant Calling Format
Typically, DNA samples are processed and sequenced using next-generation sequencing technology to generate raw sequence files. Then, the raw sequence data is aligned to generate BAM/SAM files. Next, the mutations in the genome are identified, and the results of the mutation identification are recorded in a specific file, the format of which is called VCF.
Purpose of VCF
VCF aims to provide a universal representation method for genetic variation, including single nucleotide variants (SNPs), insertions/deletions (INDELs), structural variations (SVs), etc. It was originally developed by the 1000 Genomes project to standardize the recording and sharing of genetic variation data.
SNV refers to a substitution at a single position in the genome. For example, in the reference genome, the base at a certain position is C/G, but through testing, due to individual differences or mutations, the same position has mutated to T/A. Indel includes insertions and deletions. For example, a certain position in the reference genome is CATGATGATG, but in the individual genome it is CAATGATGAT (TG deletion and ATG insertion). This is a case of deletion and insertion, which can be expressed as CA–ATGATGAT. Note that a single insertion or deletion can also be called Indel.
Broad usage across DNA and RNA sequencing workflows
In DNA sequencing, VCF files are used to store variant data, such as in Whole Genome Sequencing (WGS), Whole Exome Sequencing (WES), and Targeted Sequencing (TAS). These files are generated after variant calling and are used for subsequent analyses, such as annotation, filtering, and comparison. For example, in the analysis workflow on the Illumina platform, VCF files are used to output variant results and support further analysis steps, such as CNV analysis and TMB calculation.
While RNA sequencing is primarily utilized for analyzing gene expression, VCF files are also applicable in identifying and annotating RNA editing sites. Tools like RCARE enable the transformation of RNA-seq data into VCF format, which can then be integrated with other analytical platforms such as STAR or GATK to detect RNA editing events. Moreover, by aligning RNA-seq with corresponding DNA-seq data, researchers can compare their respective VCF outputs to uncover putative editing sites through differential analysis.
Structural Elements of a VCF File
A VCF file consists of three main parts: a metadata line, a header line, and a data line.
Meta information line: starts with “##”, contains file version, reference genome information, annotation field definition, etc.
Each VCF file has a header row, a single line, prefixed by a pound sign (#), with 8 mandatory fields separated by tabs, representing the columns of each data row. They are CHROM POS ID REF ALT QUAL FILTER INFO. If there is genotype data, a FORMAT column is declared, followed by the sample name (Sample).
The main part is the core of the VCF file, which records the specific information of each variant. Each line represents a variant record, which usually contains the following fields:
- (1)CHROM: Chromosome name.
- (2)POS: The position of the variant in the reference sequence (1-based index).
- (3)ID: variant ID, usually from the dbSNP database.
- (4)REF: Reference base sequence.
- (5)ALT: variant base sequence, which may contain multiple alternative sequences.
- (6)QUAL: Quality score, indicating the credibility of the variant.
- (7)FILTER: Filter status, indicating whether the variant passed quality control.
- (8)INFO: Additional information field containing detailed information about the variant, such as allele frequency (AF), allele depth (AD), coverage (DP), etc.
- (9)FORMAT: Format field, which defines the format of subsequent sample data.
- (10)Sample data: genotype information of each sample, including GT (genotype), AD (allele depth), DP (coverage), etc.
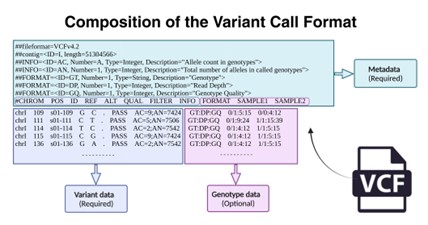
Figure 1. Composition of the standard VCF file.(Kamaraj, V et al. 2024)
Tools and File Generation of VCF
Variant callers generating VCF
GATK HaplotypeCaller: Developed by the Broad Institute, it is one of the current industry standard tools. It can improve detection accuracy by reconstructing the local map of each variant region (local de novo assembly). It supports single-sample and multi-sample joint analysis and outputs standard VCF or gVCF formats, which is suitable for high-quality, high-throughput data.
FreeBayes: A multi-sample variant detector based on Bayesian inference methods, suitable for non-human species, population sequencing and complex sample types. Supports multi-allelic loci and multi-nucleotide polymorphisms.
Samtools/BCFtools: A classic combination of variant detection tools that runs fast and is suitable for small-scale samples or scenarios with high speed requirements. Although its accuracy is slightly lower than GATK, it is lightweight and efficient.
Mutect2 (GATK component): Designed for tumor-normal paired samples, suitable for detecting low-frequency somatic mutations. It integrates a variety of filtering and correction modules to effectively eliminate false positives.
Manta: Developed by Illumina, it supports the detection of multiple types of structural variations from BAM files. It is suitable for somatic and germline samples, and takes into account both speed and accuracy.
The following is a summary list of common variant detection tools , classified by purpose, including detection type, applicable scenarios, and whether VCF files can be generated:
| Tool Name | Developer/Organization | Applicable types | Features and Benefits |
| GATK HaplotypeCaller | Broad Institute | SNP、Indel | Industry standard, high accuracy, supports gVCF mode, suitable for large-scale analysis |
| FreeBayes | Erik Garrison | SNP、Indel | Based on Bayesian model, suitable for multi-sample analysis |
| Samtools/BCFtools | Heng Li et al. | SNP、Indel | Fast operation, suitable for low-depth data |
| Mutect2(GATK) | Broad Institute | Somatic (tumor) | Designed specifically for tumor mutation detection, processing tumor-normal sample pairs |
| Manta | Illumina | Structural variation (SV) | Suitable for germline and somatic SV detection |
Compatible sequencing types
Whole genome sequencing: Tools such as GATK, bcftools, and FreeBayes are widely used for variant detection in whole genome sequencing data. For example, GATK supports the GVCF mode, which can process WGS data of multiple samples and generate high-quality VCF files for downstream analysis.
RNA sequencing: RNA-seq data is also suitable for variant detection analysis. Tools such as GATK can identify SNPs and Indels in RNA-seq data and output standardized VCF files to support subsequent functional annotation and analysis.
Long-read sequencing (PacBio, Nanopore): The above tools can also be used to process long-read sequencing data generated by platforms such as PacBio or Nanopore. Taking GATK as an example, it supports accurate variant detection of long-read data and generates high-quality VCF files to meet the analysis needs of structural variations and complex regions.
Applications of VCF in Research and Clinical Genomics
VCF files are not only a standard storage format for variant data, but also a bridge between basic research and clinical practice. In practical applications, they are used to build population genetic variation maps, study disease mechanisms, and make precision medicine decisions.
(1) Group-level database
VCF files are the basic format for building human genetic diversity databases and are widely used in population genetics, disease research, and clinical variation interpretation.
1000 Genomes Project: This project is the first international collaborative project to systematically construct a global population genetic variation map, covering more than 2,500 samples and 26 ethnic groups. By analyzing its public VCF files, researchers have discovered more than 88 million variant sites. These data have become the control baseline for many genetic studies, helping researchers to screen out high-frequency benign variants from patient VCFs and focus on rare potential pathogenic mutations.
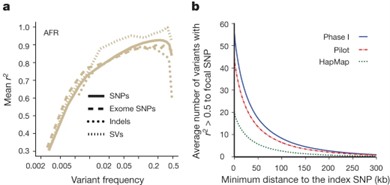
Figure 2. Implications of phase I 1000 Genomes Project data for GWAS.
gnomAD (Genome Aggregation Database): The gnomAD database integrates a large amount of whole genome and exome data, provides variant allele frequency information, and is an important tool for clinical variant interpretation. It helps researchers distinguish between pathogenic variants and benign variants by filtering out common polymorphisms and focusing on rare variants . In addition, these databases also provide basic data support for the discovery and functional research of disease-related genes, such as revealing their potential evolutionary conservation and functional impact by analyzing the geographical distribution of low-frequency variants.
(2) Precision Oncology
In cancer research and personalized medicine, VCF files are widely used to identify treatment-related mutation information. Specific applications include:
- Targeted therapy mutation screening : VCF files can detect mutations associated with sensitivity to targeted drugs, such as EGFR, BRAF, and KRAS. The detection of these mutations is crucial to guide patients in choosing appropriate targeted therapy options .
- Identification of drug resistance mechanisms: By longitudinally analyzing VCF data at multiple time points, we can track the evolution of the tumor genome and identify secondary mutations associated with treatment resistance. This analysis helps develop new treatment strategies and optimize existing therapies.
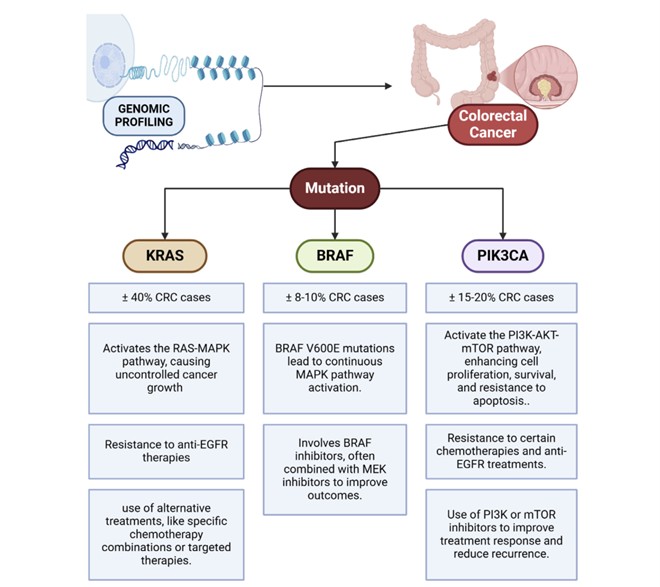
Figure 3. Identification of KRAS, BRAF, and PIK3CA mutations in colorectal cancer. (Muradi Muhar, A. et al. 2025)
File Handling and Compression Strategies
When processing large-scale genomic data, VCF files face challenges in storage and processing efficiency due to their large file size. To solve these problems, the following strategies are usually adopted:
VCF files are often compressed using bgzip, a tool based on the GZIP algorithm that is able to significantly reduce file size while preserving data integrity. For example, bgzip can compress VCF files into .gz format, saving storage space.
Use the Tabix tool to index the compressed VCF file to quickly access data in a specific region. Index files (such as .tbi) allow users to efficiently retrieve information about a specified chromosome location or SNP site. This indexing method is particularly suitable for fast queries of large-scale data sets.
For scenarios that require further optimization, VCF files can be converted to binary format (BCF), which is more compact and faster to load than VCF. For example, the bcftools tool supports converting VCF files to BCF format and enables efficient random access through indexes.
In conclusion, the VCF has become an indispensable tool in modern genomics research and clinical applications as the standard format for representing genetic variants. Its flexible and extensible structure accommodates a wide range of sequencing data and variant types, while allowing seamless integration with diverse variant-calling tools to facilitate data sharing and cross-platform analysis. With the advancement of large-scale population studies and precision medicine, VCF files will continue to play a pivotal role in variant database construction, disease mechanism investigation, and the development of personalized therapeutic strategies. Looking ahead, the integration of more efficient data compression and indexing technologies is expected to further enhance the processing efficiency and standardization of variant data.
References:
- Kamaraj, V., & Sinha, H. (2024). SCI-VCF: a cross-platform GUI solution to summarize, compare, inspect and visualize the variant call format. NAR genomics and bioinformatics, 6(3), lqae083. https://doi.org/10.1093/nargab/lqae083
- 1000 Genomes Project Consortium, et al. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature, 491(7422), 56–65. https://doi.org/10.1038/nature11632