
 Sample Submission Guidelines
Sample Submission Guidelines
metatranscriptomics integrates expertise from multiple disciplines and techniques to examine the transcriptional products (mainly mRNA) of entire biological communities in specific environments. Moving beyond the constraints of traditional microbiological studies, it uses high-throughput sequencing and advanced bioinformatics to expose comprehensive gene expression patterns in microbial communities. This approach has transformed research in microbial ecology, environmental science, and medicine, helping clarify microbial roles and functions while solving real-world challenges.
This article highlights metatranscriptomic case studies from five representative domains: marine environments, the human gut, soil, wastewater treatment systems, and polar regions. By detailing research goals, methods, and findings, we demonstrate the benefits and promise of metatranscriptomics for uncovering microbial community dynamics, functional mechanisms, and interactions with the environment, offering guidance for future studies.
Introduction to Metatranscriptomics
Metatranscriptomics is a field that examines the transcriptional products (especially mRNA) of entire biological communities in defined environmental settings. It transcends the limitations of traditional microbiology, which often centers on isolated species, by profiling gene expression in microbial communities holistically. This enables insights into microbial functional activities, metabolic networks, and interaction mechanisms across diverse habitats.
By leveraging high-throughput sequencing to capture transcriptional information from environmental microorganisms and integrating this with bioinformatics, metatranscriptomics enables the discovery and interpretation of extensive datasets. This innovative method has reshaped research in microbial ecology, environmental science, and medicine. It reveals the functions and ecological roles of microbes and offers novel solutions for tackling environmental pollution, disease prevention, and control.
Case Study 1: Response of Marine Microbial Communities to Oil Pollution
Marine microorganisms are integral to marine ecosystems. Understanding how they respond to environmental shifts and their underlying functional mechanisms is vital for crafting effective marine management strategies.
Title: Marine Microeukaryote Metatranscriptomics: Sample Processing and Bioinformatic Workflow Recommendations for Ecological Applications
Journal: Frontiers in Marine Science
Impact Factor: 3.7 (2022 JCR)
Publication Date: June 28, 2022
DOI: 10.3389/fmars 2022.867007(no follow)
Sample Selection: Forty-four seawater samples (0.8–200 µm particle size) were collected along a transect in the western Atlantic Ocean, covering the surface to a 350 m vertical profile and areas with varying nutrient levels.
Research Techniques: Metatranscriptomics
Background: Traditional 18S rRNA amplicon sequencing struggles to capture real-time gene expression and metabolic states of microeukaryotes in natural environments; metatranscriptomics fills this gap, but standardized protocols for sample processing and bioinformatics are lacking.
Objective: To establish a standardized microeukaryote metatranscriptomics protocol, from field sampling to downstream ecological applications, to accurately assess the functional responses of marine microeukaryotic communities to environmental changes.
Research Approach and Results: The research team targeted marine microeukaryote communities, using McLane pumps or AUVs to filter up to 350 L of seawater in situ. They preserved samples within 30 minutes and extracted total RNA with a combined thermal lysis and silica bead method, followed by DNase I treatment to remove DNA. The team applied quality control with Trimomatic, parallel assembly with Trinity/MEGAHIT/TransABySS, quantification with Salmon, and functional annotation with eggNOGmapper in their bioinformatics workflow. They introduced synthetic mRNA internal standards to estimate absolute transcript copy numbers per liter of seawater. For the first time, they resolved 77,438 protein families and 3.1 million spectral counts across the Atlantic transect and revealed differences in photosynthetic gene expression among diatoms and dinoflagellates along nutrient gradients. This approach provides high-resolution data for studying phytoplankton community function.
Impact: This standardized protocol has been incorporated into the joint ecological observation network of the NSF and NOAA, significantly enhancing the accuracy, comparability, and high-throughput capabilities of marine microeukaryotic community function research. It offers a new tool for assessing climate change, eutrophication, and biogeochemical cycles.

Utilizing metatranscriptomics to investigate marine microbial communities (Cohen et al., 2022)
Case Study 2: The Relationship Between Human Gut Microbiota and Inflammatory Bowel Disease (IBD)
Inflammatory Bowel Disease (IBD) is a persistent inflammatory disorder of the intestines. Its pathogenesis is complex, involving genetic, environmental, and immune factors. Recent studies increasingly emphasize the key role of the human gut microbiota in IBD initiation and progression.
Title: Mining the Microbiome and Microbiota-Derived Molecules in Inflammatory Bowel Disease
Journal: International Journal of Molecular Sciences
Impact Factor: 5.9 (2021 JCR)
Publication Date: October 18, 2021
DOI: 10.3390/ijms222011243
Sample Selection: The study included stool, serum, and intestinal biopsy samples from 535 IBD patients (including Crohn’s Disease (CD) and Ulcerative Colitis (UC)) and healthy controls. First-degree relatives of IBD patients were also matched as a high-risk group for longitudinal follow-up.
Research Techniques: Metatranscriptomics
Background: Traditional 16S rRNA sequencing can only describe "what is present," whereas metatranscriptomics captures real-time gene expression of gut microbiota, revealing their functional roles in inflammation.
Objective: To utilize metatranscriptomics to mine the human gut microbiota and their derived active molecules, providing new targets for early diagnosis, prognosis assessment, and precision treatment of IBD.
Research Approach and Results: The research team implemented a metatranscriptomics strategy on stool samples from 535 IBD patients and healthy controls. They immediately flash-frosted collected samples in liquid nitrogen; after rRNA depletion, NovaSeq PE150 sequencing produced >2×10⁷ reads per sample. The team used Salmon for quantification and eggNOG/KEGG annotations to identify inflammation-related functional genes. Their results showed a significant decrease in transcriptional activity of butyrate-producing bacteria such as Faecalibacterium prausnitzii and Roseburia intestinalis in patients’ intestines, while Ruminococcus gnavus and E. coli were upregulated. They observed that aromatic amino acid metabolic pathway activity correlated with indole-3-acetic acid and secondary bile acid levels detected by LC-MS/MS. These metabolites inhibited Th17 inflammation via AHR/FXR. The random forest model built from these data achieved an AUC of 0.87 in predicting IBD activity in the validation cohort. The researchers established indole pathway genes as early biomarkers for treatment response, laying the foundation for microbiota metabolite-targeted drugs and personalized microbial interventions.
Impact: The team became the first to apply metatranscriptomics to a large-scale IBD population, clarifying the direct link between microbial functional genes and host inflammation. Their work provides both a technological and biomarker foundation for developing microbiota metabolite-targeted drugs and personalized microbial interventions.
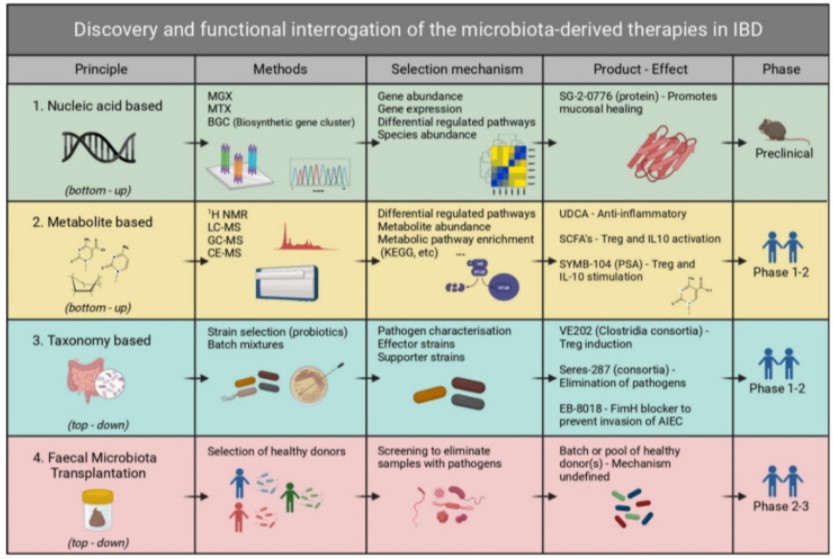
Metatranscriptomics uncovers alterations in microbial functions linked to disease development (Bekkers et al., 2021)
Case Study 3: Functional Changes in Soil Microbial Communities Under Agricultural Fertilization Management
In agriculture, proper fertilizer application is crucial for improving crop yield and quality. Yet, long-term misuse can disturb soil microbial structure and function, degrading soil fertility and damaging the ecosystem.
Title: Comparative Metatranscriptome Analysis Revealed Broad Response of Microbial Communities in Two Soil Types, Agriculture versus Organic Soil
Journal: Journal of Genetic Engineering & Biotechnology
Impact Factor: 2.3 (2020 JCR)
Publication Date: October 28, 2019
DOI: 10.1186/s4314101900063(no follow)
Sample Selection: Agricultural soil (M1) from Punjab, India, with long-term application of chemical fertilizers/pesticides, and adjacent organically managed soil (O1), with five replicate sampling points each.
Research Techniques: Metatranscriptomics
Background: Agricultural fertilization management alters soil physicochemical properties, driving changes in microbial community structure and functional gene expression. However, real-time activity information is lacking.
Objective: To compare the functional differences in soil microbial communities under two management modes using metatranscriptomics and identify molecular markers indicative of fertilization/pollution stress.
Research Approach and Results: The research team collected five replicate soil samples each from agricultural land in Punjab, India, with long-term chemical fertilizer/pesticide use, and adjacent organically managed land. They immediately flash-frosted the samples in liquid nitrogen for RNA extraction. After assembly, quantification, and GO/KEGG annotation, they found that transcripts from Proteobacteria, Ascomycota, and Firmicutes dominated in agricultural soil. In contrast, transcripts from Cyanobacteria and Actinobacteria showed high expression in organic soil. They saw that functional genes such as copper-binding proteins, MFS transporters, and aromatic hydrocarbon degradation dioxygenases were significantly upregulated in agricultural soil, with enhanced activities of nitrification, ammonification, and alternative carbon fixation pathways. The team also revealed, for the first time, that archaea contribute more to nitrification under pesticide/heavy metal stress than bacteria. Finally, they identified a set of transcripts that can real-time indicate fertilization pressure, providing functional gene markers for precision fertilization and soil health diagnosis.
Impact: The study applied metatranscriptomics to agricultural fertilization management, offering real-time functional gene markers for precision fertilization, soil health diagnosis, and pollution bioremediation.
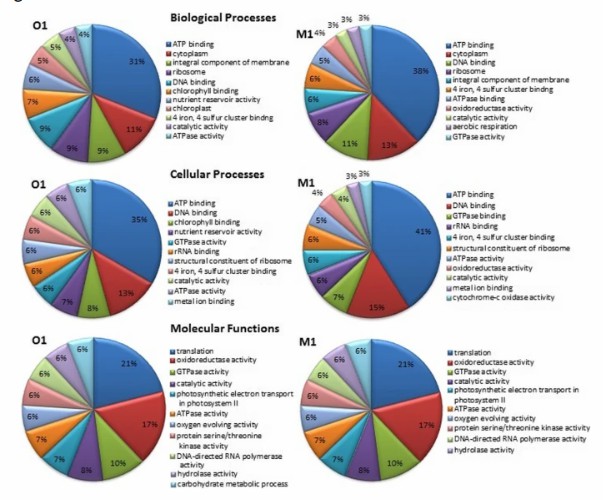
Employing metatranscriptomics for the study of soil microbial communities (Sharma et al., 2019)
Case Study 4: Functional Analysis of Microbial Communities in Wastewater Treatment Systems
Wastewater treatment is essential to safeguard water resources and prevent pollution, with microorganisms playing a critical role. Applying metatranscriptomics to study microbial communities in treatment systems deepens our understanding of microbial degradation and informs process optimization.
Title: Metatranscriptomes of Activated Sludge Microbiomes from Saline Wastewater Treatment Plant
Journal: Scientific Data
Impact Factor: 8.5
Publication Date: February 26, 2025
DOI: 10.1038/s4159702504682w (no follow)
Sample Selection: The study collected 32 samples from a high-salinity activated sludge system (SWWTP) associated with a seawater desalination plant, covering aeration tanks, anoxic tanks, and secondary sedimentation tanks, with salinity levels ranging from 20 to 35 g/L. Conventional municipal wastewater treatment plants were also sampled as controls to compare the impact of salinity on microbial communities.
Research Techniques: Metatranscriptomics
Background: While 16S rRNA sequencing can reveal species composition, it fails to capture active metabolism. Metagenomics can predict potential functions, but cannot distinguish expressed genes from silent ones. Evidence on how high-salinity environments affect the functionality of activated sludge microbial communities at the transcriptional level is still lacking.
Objective: To utilize metatranscriptomics to dissect the real-time functions and core metabolic pathways of microbial communities in high-salinity activated sludge, providing molecular insights for optimizing high-salinity wastewater treatment processes.
Research Approach and Results: The research team rapidly filtered large volumes of high-salinity activated sludge and flash-frosted it in liquid nitrogen within 30 minutes. After total RNA extraction and rRNA removal, they sequenced each sample on NovaSeq PE150, yielding >2×10⁷ reads per sample. The team applied Trimomatic for quality control, MEGAHIT for assembly, Salmon for quantification, and KEGG/SEED for functional annotation. Their analysis revealed, for the first time under salinity gradients, that Pseudomonadota became the dominant active group, with significantly upregulated genes for nitrate reduction, organic substrate degradation, and salt-tolerant osmoregulation. Thermodesulfobacteriota was enriched and activated sulfate reduction pathways. They also found that core functional genes for nitrogen, phosphorus, and sulfur cycling showed marked differences from those in municipal wastewater plants, offering real-time transcriptional evidence for optimizing high-salinity wastewater processes and discovering novel functional microbial resources.
Impact: This study successfully applied metatranscriptomics to high-salinity wastewater treatment systems, offering real-time evidence of salinity-driven changes in functional gene expression and laying a data and methodological foundation for optimizing high-salinity activated sludge processes and discovering new functional microbial resources.
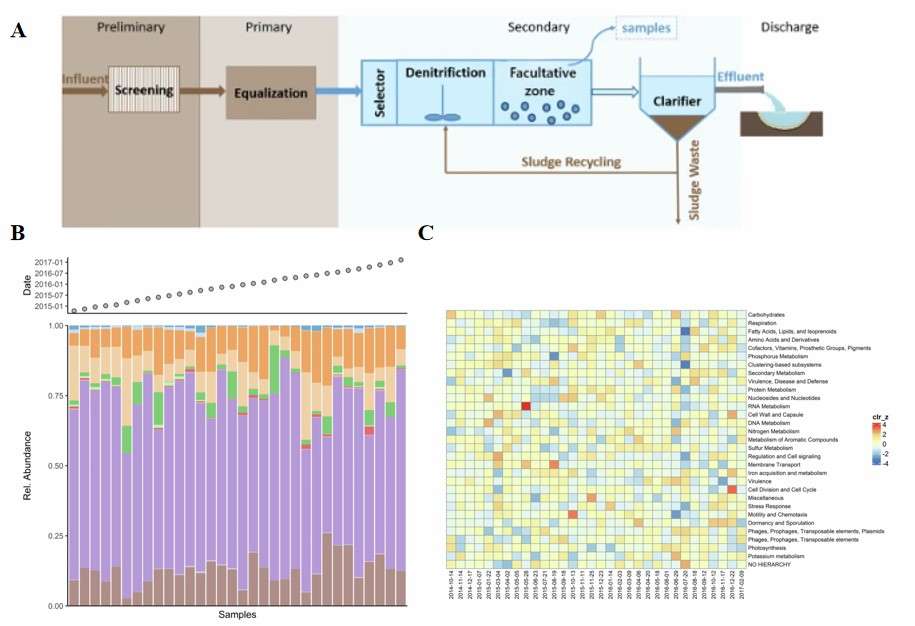
Applying metatranscriptomics to dissect the functional characteristics of microbial communities in wastewater treatment systems (Mahajna et al., 2025)
Conclusion
By closely examining four metatranscriptomic case studies across diverse fields, we gain a deeper appreciation for the power and broad applicability of metatranscriptomics in revealing microbial community functions. Whether in marine environments, the human gut, soil, wastewater treatment, or polar regions, metatranscriptomics provides comprehensive gene expression insights. This advances our understanding of microbial adaptation, functional roles, and environmental interactions.
Learn More
- Overview of Metatranscriptomic Sequencing: Principles, Workflow, and Applications
- 16S rRNA Metatranscriptomics: Workflow, Applications, and Challenges
- 18S rRNA Metatranscriptomics: Principles, Applications, and Challenges
- Metagenomics vs Metatranscriptomics: Unveiling Microbial Communities & Gene Expression Dynamics
References
- Cohen NR, Alexander H, Krinos AI, Hu SK, Lampe RH. "Marine Microeukaryote Metatranscriptomics: Sample Processing and Bioinformatic Workflow Recommendations for Ecological Applications." Front Mar Sci. 2022;9:867007. https://doi.org/10.3389/fmars.2022.867007.
- Bekkers M, Stojkovic B, Kaiko GE. "Mining the Microbiome and Microbiota-Derived Molecules in Inflammatory Bowel Disease." International Journal of Molecular Sciences. 2021;22(20):11243. https://doi.org/10.3390/ijms222011243.
- Sharma PK, Sharma V, Sharma S, et al. "Comparative metatranscriptome analysis revealed broad response of microbial communities in two soil types, agriculture versus organic soil." J Genet Eng Biotechnol. 2019;17:6. https://doi.org/10.1186/s4314101900063.
- Mahajna A, Geurkink B, Gacesa R, et al. "Metatranscriptomes of activated sludge microbiomes from saline wastewater treatment plant." Sci Data. 2025;12:348. https://doi.org/10.1038/s4159702504682w.