Introduction
Exosomal RNA-Seq is not just a variant of standard transcriptomics—it is a precision workflow built for a uniquely complex biological substrate. Exosomes, a class of extracellular vesicles ranging from 30–150 nm in diameter, carry RNA cargo that reflects the physiological state of their cell of origin. This makes exosomal RNA (exoRNA) an invaluable resource for understanding cell–cell communication, disease mechanisms, and treatment responses in a non-invasive manner. However, unlocking these insights requires careful adaptation at every stage of the RNA sequencing pipeline.
Unlike cellular RNA, exoRNA is often scarce, fragmented, and diverse in composition. It includes small non-coding RNAs such as microRNAs (miRNAs), as well as lncRNAs, circular RNAs, and degraded mRNA fragments. These RNA populations are often present in nanogram quantities, requiring ultra-sensitive workflows and rigorous contamination control to distinguish true exosomal signals from background cell-free RNA (cfRNA). In addition, variability in exosome content across plasma, urine, and cell culture supernatants demands context-specific handling and analysis strategies.
For researchers pursuing biomarker discovery, pathway exploration, or translational research in vitro or in vivo, a dedicated exosome RNA-Seq workflow ensures that biologically meaningful results are not lost to poor preparation, inappropriate tools, or misinterpretation. From sample selection and isolation to sequencing and interpretation, each step must be optimized for the nuances of extracellular RNA biology.
Related Reading:
What Is Exosome RNA Sequencing and Why It Matters in Modern Biology
Sample Selection and Preparation
A successful exosomal RNA-Seq project starts long before sequencing—it begins with choosing the right sample type and applying rigorous handling practices. Since exosomes are present in nearly all biofluids, researchers have multiple input options, but each comes with its own technical trade-offs. Selecting the most appropriate sample type based on project goals, RNA yield, and processing feasibility is critical to downstream success.
Common Biofluid Sources for Exosome RNA-Seq
| Sample Type |
Suitability |
Key Considerations |
| Plasma (EDTA tubes) |
★★★★☆ (Preferred) |
High exosome concentration; avoid heparin due to downstream PCR/NGS inhibition. |
| Serum |
★★★☆☆ |
Common but may contain platelet-derived vesicles and clotting-induced variability. |
| Urine |
★★★☆☆ |
Non-invasive; lower RNA yield; quick processing needed to prevent degradation. |
| Saliva |
★★☆☆☆ |
Easy to collect but highly variable in exosome content and RNase activity. |
| Cell Culture Supernatant |
★★★★★ (Ideal for mechanistic studies) |
Clean background, controllable conditions, and vesicle uniformity for profiling. |
Quality Control Considerations
Regardless of sample type, rigorous pre-analytical practices are essential to protect RNA quality:
Time-to-processing: Process plasma/serum within 2 hours of collection to avoid cfRNA contamination from lysed blood cells.
Centrifugation: Use at least two-step centrifugation to remove debris and apoptotic bodies before exosome isolation.
Storage: Aliquot and freeze samples at −80°C to avoid freeze–thaw degradation.
Volume: Ensure sufficient starting material. For plasma, 1–2 mL is typically required to obtain enough RNA for sequencing.
Choosing the right input isn't just about convenience—it directly impacts RNA integrity, library complexity, and ultimately the resolution of your biological insights.
Related Reading: How to Prepare Samples for Exosome RNA Sequencing: A Step-by-Step Guide
Exosome Isolation and RNA Extraction Options
Exosome isolation and RNA extraction form the cornerstone of any successful exosomal RNA-Seq workflow. These upstream steps directly influence the quality, purity, and interpretability of your sequencing data. Because exosomal RNA is low in abundance and highly susceptible to contamination (especially from cell-free RNA or protein complexes), the methods used for vesicle enrichment and RNA purification must be carefully selected.
Common Exosome Isolation Methods
Each method offers trade-offs between purity, scalability, cost, and ease of use:
| Method |
Best For |
Pros |
Limitations |
| Ultracentrifugation |
High-purity research applications |
Gold standard; good vesicle enrichment |
Time-consuming; requires expensive equipment |
| PEG Precipitation |
High-throughput, lower-budget projects |
Scalable; easy to implement |
Lower purity; risk of co-precipitating contaminants |
| Affinity-Based Kits |
Targeted exosome populations (e.g., CD63+) |
Specific and fast; low sample volume required |
May bias vesicle subtypes; costly |
| Size-Exclusion Chromatography (SEC) |
Plasma or serum samples |
Gentle on vesicles; reduces protein carryover |
Requires additional steps for concentration |
For sample types like plasma or urine, SEC followed by ultrafiltration can offer a good balance of purity and recovery. For cell culture media, ultracentrifugation remains a preferred approach due to low background noise.
RNA Extraction from Isolated Exosomes
RNA yield from exosomes is typically in the nanogram range, and the RNA population includes a high proportion of small RNAs. The choice of extraction method should prioritize:
Retention of small RNAs (e.g., miRNAs)
Avoidance of contaminants like phenol, guanidinium, or residual proteins
Buffer compatibility with downstream library preparation (e.g., no EDTA)
Popular methods include:
- Phenol-free column-based kits optimized for small RNAs (e.g., Norgen, Qiagen miRNeasy)
- Magnetic bead–based kits, which offer automation potential and cleaner RNA in low-volume settings
- TRIzol-based methods (less preferred due to reproducibility and toxicity concerns)
Quality Control Metrics to Target:
- RNA concentration ≥1 ng/µL
- Total RNA yield ≥20 ng
- OD260/280 between 1.8–2.2; OD260/230 ≥2.0
- RIN ≥6.5 if using total RNA; Bioanalyzer traces recommended
Related Reading: Choosing the Right RNA Isolation Method for Exosomes
Careful attention to these steps ensures that your exosomal RNA is not only sufficient in quantity but also free of inhibitors that can compromise library construction and sequencing fidelity.
Library Construction – miRNA vs Total RNA Strategies
Library preparation is a pivotal step in the exosome RNA-Seq workflow, dictating both the sensitivity and the specificity of your results. Because exosomes carry a complex mixture of RNA species—including small RNAs like miRNAs and fragmented long RNAs such as lncRNAs and mRNA remnants—the choice of library strategy must align with your biological question and sample constraints.
miRNA Library Construction
Exosomal miRNAs are short (~22 nucleotides), abundant, and often central to regulatory signaling. Their small size requires a dedicated workflow that includes:
Sequential adapter ligation: Specialized 3′ and 5′ adapters are ligated to the ends of small RNAs to enable downstream amplification.
Size selection: A purification step selectively enriches small RNAs (typically 18–30 nt), removing tRNA fragments and other non-target RNAs.
Molecular indexing (optional): Some workflows incorporate sequence tags during library prep to track unique RNA molecules and reduce amplification bias.
Reverse transcription and amplification: The adapter-ligated RNAs are converted into cDNA and amplified by PCR.
For exosomal RNA samples, which are often low-input and partially degraded, protocols designed to minimize adapter-dimer formation and amplification bias are preferred.
Total RNA Library Construction (for lncRNA, mRNA fragments, and other long RNAs)
If the aim is to investigate coding or long non-coding RNA fragments in exosomes, total RNA library preparation is the appropriate path. These workflows typically involve:
rRNA removal: Ribosomal RNA is depleted to enrich for non-rRNA content. This is essential, as poly(A) tails are often missing or truncated in vesicle-derived RNA.
Fragmentation (optional): Usually skipped for exosomal RNA since vesicle-contained transcripts are already degraded into shorter fragments.
Random priming and reverse transcription: RNA is converted into cDNA using primers that cover a broad range of transcript types.
Strand-specific amplification: Maintains transcript directionality, which aids in downstream annotation and interpretation.
When working with very limited RNA amounts, ensure the protocol is compatible with low-input workflows (e.g., nanogram scale or below).
Comparative Summary
| Parameter |
Small RNA Workflow |
Total RNA Workflow |
| Target RNA Types |
miRNA, piRNA, siRNA |
lncRNA, mRNA fragments |
| Fragmentation Step |
Not required |
Optional (often skipped) |
| rRNA Removal |
Not applicable |
Required |
| Size Selection |
Mandatory (~18–30 nt) |
Not typically required |
| Strand Information |
Sometimes retained |
Usually retained |
| Input Amount (typical) |
1–10 ng |
10–100 ng |
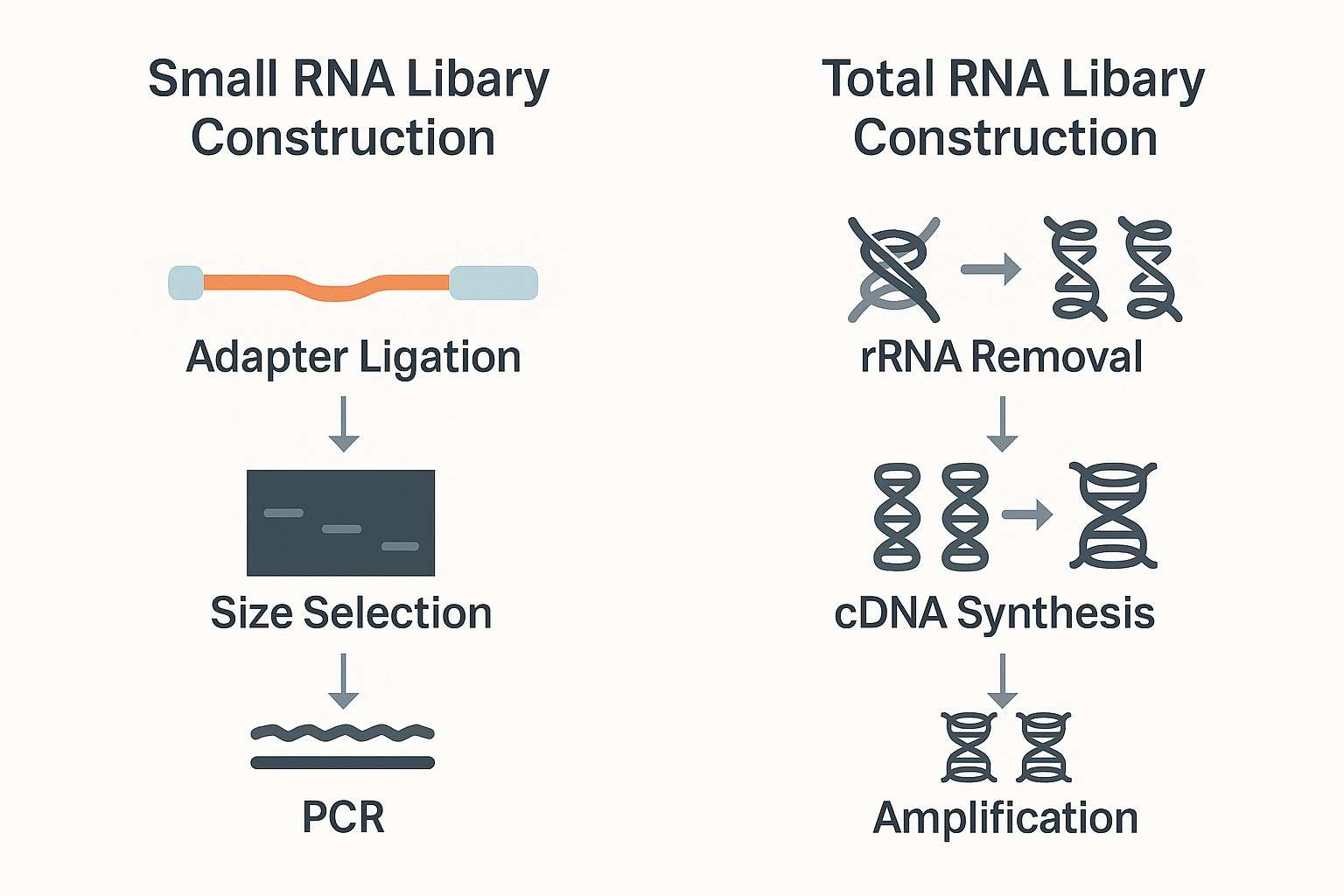 Comparison of small RNA and total RNA library preparation workflows for exosomal RNA-Seq.
Comparison of small RNA and total RNA library preparation workflows for exosomal RNA-Seq.
Once your library prep strategy is finalized, the next decision point is choosing the right sequencing platform and read parameters to match your study goals.
Sequencing Platforms and Read Parameters
Once libraries are prepared, the next critical decision is selecting appropriate sequencing parameters. Exosomal RNA, especially from low-input or small RNA-enriched libraries, demands optimized settings to ensure coverage, specificity, and interpretability. While most workflows use short-read sequencing platforms, the ideal configuration depends on the library type and the biological question being asked.
Recommended Sequencing Platforms
Short-read next-generation sequencing (NGS) platforms—particularly those offering high throughput and high base-calling accuracy—remain the gold standard for most exosomal RNA-Seq projects.
| Platform Type |
Use Case |
Advantages |
| High-throughput short-read (e.g., Illumina) |
Small RNA profiling, total RNA quantification |
High accuracy, multiplexing support, established workflows |
| Long-read (e.g., nanopore, PacBio) |
Isoform discovery, circRNA, RNA modifications |
Captures full-length transcripts, supports novel isoform detection |
For most exosome RNA-Seq studies, Illumina-style short-read platforms are preferred due to their accuracy and compatibility with small-RNA-focused workflows.
Sequencing Parameters by Library Type
| Library Type |
Recommended Read Length |
Read Type |
Depth per Sample |
| miRNA/small RNA |
50 bp single-end |
Single-end (SE) |
≥5–10 million reads |
| Total RNA (mRNA/lncRNA fragments) |
75–150 bp paired-end |
Paired-end (PE) |
≥20 million reads |
Single-end reads are sufficient for short RNA species like miRNAs.
Paired-end reads improve alignment quality and isoform discrimination in total RNA workflows.
Read depth affects sensitivity. For biomarker discovery or low-abundance transcripts, deeper sequencing is advised.
Quality Control During Sequencing
Ensure the platform provides:
- High Q30 scores (>85% of reads)
- Low index-hopping (especially important for multiplexed small RNA libraries)
- Balanced GC-content representation
- Cluster passing filters (≥80%)
Selecting the right sequencing configuration helps maximize signal-to-noise and ensures your libraries yield usable, reproducible data—whether you're profiling miRNAs or reconstructing exosomal transcript fragments.
Bioinformatics Workflow – From Raw Reads to Expression Matrix
The computational pipeline is as important as the wet lab workflow in exosomal RNA-Seq. Given the small size, low input, and diverse RNA types in extracellular vesicles, standard RNA-Seq workflows must be adapted. This section outlines a typical step-by-step data processing pipeline tailored to exosomal RNA—whether you're profiling miRNAs, lncRNAs, or mRNA fragments.
Step 1: Quality Control (QC) of Raw Reads
Tools: FastQC, MultiQC
Purpose: Assess per-base quality, GC content, adapter contamination, and read duplication.
- Small RNA libraries often contain adapter dimers and short reads; trimming is essential.
- Flagged issues like low base quality or adapter contamination must be resolved before mapping.
Step 2: Adapter Trimming and Filtering
Tools: Cutadapt, Trimmomatic
- Remove sequencing adapters, low-quality bases (Q<20), and short reads (<15 bp for miRNAs).
- For total RNA libraries, ensure ribosomal sequences or poly(A) artifacts are trimmed if not removed experimentally.
Step 3: Mapping to Reference Genome or Transcriptome
miRNA Mapping:
- Aligner: Bowtie, STAR (with miRNA mode)
- Reference: Mature miRNAs from miRBase
Total RNA Mapping:
- Aligner: STAR or HISAT2
- Reference: GENCODE, Ensembl, or species-specific transcriptome
- Exosomal RNA may contain degraded or fragmented transcripts—permissive mapping parameters help capture true alignments.
Step 4: Quantification
miRNA:
- Use featureCounts, HTSeq, or specialized tools like miRDeep2 or sRNAbench to count miRNA reads.
Total RNA:
- For lncRNA/mRNA, use featureCounts, Salmon, or RSEM for transcript-level abundance estimates.
Step 5: Normalization
Tools: DESeq2, edgeR, or TPM/CPM calculation
- Normalize to account for sequencing depth and library composition.
- Log-transformed counts often improve downstream analysis.
Output: Expression Matrix
The final matrix is a table of gene/miRNA IDs vs. normalized read counts—ready for downstream functional analysis or differential expression.
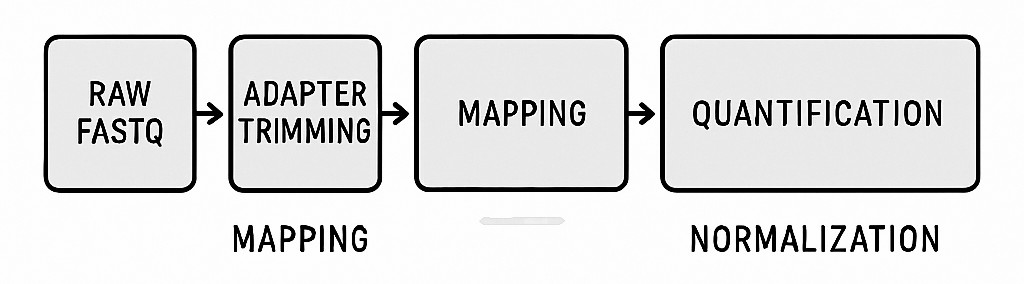 Workflow diagram of the exosome RNA-Seq bioinformatics pipeline from raw reads to expression matrix.
Workflow diagram of the exosome RNA-Seq bioinformatics pipeline from raw reads to expression matrix.
This bioinformatics workflow is the computational backbone of exosomal RNA-Seq. It ensures that even picogram-level transcripts are interpreted with scientific rigor and statistical reliability.
Functional and Pathway Analysis
Once your exosomal RNA-Seq dataset is mapped and normalized, the next step is to extract biological meaning. Functional and pathway analysis helps contextualize differential expression results, revealing how exosome-mediated signals might influence cellular pathways, immune responses, or intercellular communication. These downstream tools are particularly valuable when studying small RNAs like miRNAs, which often regulate gene expression indirectly through target mRNA networks.
Gene Ontology (GO) and Pathway Enrichment
Tools:
- GOseq, clusterProfiler (R packages)
- g:Profiler (web-based tool)
- KEGG Mapper
Applications:
- Identify overrepresented biological processes (e.g., apoptosis, angiogenesis)
- Classify exosomal mRNA or lncRNA targets into canonical pathways
- Explore vesicle involvement in stress response, immune modulation, or developmental signaling
Example:
A differentially expressed set of lncRNAs from exosomes isolated from hypoxia-conditioned cells may enrich pathways like "HIF-1 signaling" or "VEGF receptor activity"—highlighting a functional link between vesicle content and environmental stress adaptation.
miRNA Target Prediction and Network Analysis
Tools:
Use Cases:
- Predict target genes of upregulated or downregulated miRNAs
- Build regulatory networks showing how exosomal miRNAs may silence pathways in recipient cells
- Perform enrichment on predicted targets (e.g., GO, KEGG, Reactome)
Considerations for Exosomal RNA
- Low RNA abundance means fewer detectable targets—focus on consistent, moderate-expression transcripts rather than only extreme fold changes.
- Fragmented mRNAs may not map to canonical 3′ UTRs; interpret results conservatively.
- Vesicle-specific pathways (e.g., endosomal transport, exocytosis) may be especially relevant when analyzing cargo profiles.
For exploratory studies, combining miRNA–mRNA correlation with pathway analysis offers a systems-level perspective on exosome function.
Functional analysis transforms sequencing output from a table of counts into biological insight—mapping your results onto the cellular processes that matter most for your research model.
Data Interpretation and Biological Context
Even with high-quality sequencing and robust analysis, biological insight depends on correct interpretation. Exosomal RNA data presents unique challenges that differ from cellular transcriptomes—both in composition and contextual relevance. Misreading the data can lead to incorrect conclusions or missed discoveries.
Common Pitfalls in Exosomal RNA Data Interpretation
| Pitfall |
Impact |
| Overinterpreting low-abundance transcripts |
Small fold changes in near-background levels can be statistically unreliable. |
| Ignoring cfRNA contamination |
Cell-free RNA may dominate poorly handled samples and obscure true vesicle signals. |
| Misattributing fragmented mRNAs as full-length |
Many exosomal mRNAs are degraded; annotations must consider fragment size. |
| Assuming miRNA changes equate to function |
Expression ≠ activity—miRNA function depends on target availability and cellular context. |
| Skipping normalization |
Raw read counts can mislead, especially in low-input or uneven-depth samples. |
Proper normalization (e.g., using size factors, spike-ins, or appropriate scaling methods) is essential for accurate cross-sample comparison.
Practical Tips for Interpreting Exosomal RNA Results
Validate with biological replicates – One sample rarely tells the full story. Pooling and replication reduce bias.
Cross-reference expression with functional data – Does a miRNA's target gene network support its proposed role?
Use vesicle databases for context – Confirm RNA species with Vesiclepedia or ExoCarta to ensure they're vesicle-enriched.
Visualize with care – Use heatmaps, volcano plots, and miRNA–target networks to detect patterns, but interpret statistically.
Integrate metadata – Clinical metadata (e.g., timepoints, treatments) helps contextualize biological trends in expression.
When to Seek Expert Support
- Low-input or degraded samples
- Conflicting results between replicates
- Need for cross-species analysis
- Custom pathway enrichment or cross-omics integration
If you're unsure about your data's reliability or significance, early consultation with a bioinformatics specialist can prevent costly missteps.
Related Reading: Interpreting Exosomal RNA Sequencing Data
Case Study Example – Exosomal RNA-Seq in Action
A 2024 study published in BMC Cancer by Huang et al. explored the potential of plasma-derived exosomal RNA as non-invasive biomarkers for hepatocellular carcinoma (HCC) detection. The researchers identified three genes—CDK1, FEN1, and PCNA—that were significantly upregulated in the plasma exosomes of HCC patients compared to non-HCC individuals, including those with hepatitis B virus (HBV) infection and healthy controls.
Study Highlights:
Data Integration: The study analyzed multiple public RNA-seq datasets from liver tissues (normal, peritumoral, and tumor) to identify differentially expressed genes associated with HCC.
Biomarker Identification: Through pathway enrichment and protein–protein interaction network analyses, ten key genes were initially identified. Further evaluation narrowed this to three genes—CDK1, FEN1, and PCNA—that showed significant elevation in plasma exosomes of HCC patients.
Diagnostic Model: A multi-layer perceptron (MLP) model was developed using the expression levels of these three genes. The model demonstrated strong diagnostic performance, with an area under the curve (AUC) of 0.85 in the training set and 0.84 in the test set, after adjusting for sex and age.
Implications for Research:
This study underscores the utility of exosomal RNA-Seq in identifying non-invasive biomarkers for HCC. The approach of integrating tissue RNA-seq data with plasma exosomal RNA profiles, coupled with machine learning techniques, provides a robust framework for biomarker discovery and validation.
Reference:
Huang H, Zhang M, Lu H, et al. Identification and evaluation of plasma exosome RNA biomarkers for non-invasive diagnosis of hepatocellular carcinoma using RNA-seq. BMC Cancer. 2024;24(1):1552. doi: 10.1186/s12885-024-13332-0
CD Genomics – One-Stop Exosomal RNA-Seq Service
At CD Genomics, we offer an end-to-end Exosomal RNA Sequencing service tailored for research teams across academia, biotech, and pharmaceutical R&D. Our workflow is built on a deep understanding of the unique challenges associated with extracellular RNA analysis and optimized for low-input, vesicle-derived RNA.
What's Included in Our Service:
1. Technical Consultation
We start by reviewing your research goals, sample type, and RNA quality to ensure project feasibility and guide input requirements.
2. Quality Control (QC) and RNA Assessment
Each submitted RNA sample undergoes rigorous QC, including:
- RNA Integrity Number (RIN) or small RNA profile
- OD260/280 and OD260/230 ratio checks
- Quantification via Qubit and Bioanalyzer/TapeStation
3. Library Construction
We support both small RNA (e.g., miRNAs) and total RNA (e.g., mRNA fragments, lncRNA) workflows. All steps are carefully optimized for:
- Adapter ligation efficiency
- Fragment recovery
- Amplification bias control
4. High-Throughput Sequencing
Utilizing Illumina-compatible platforms, we deliver:
- Single-end or paired-end reads
- Customizable read lengths and depths (5M–30M reads/sample)
- Barcode demultiplexing and format conversion
5. Bioinformatics Analysis
Our bioinformatics team processes raw data through validated pipelines for:
- Adapter trimming and quality filtering
- Mapping to the reference genome or miRNA database
- Expression quantification (counts, TPM/RPM)
- Differential expression analysis (DESeq2, edgeR)
- Functional enrichment (GO, KEGG, miRNA–target prediction)
6. Publication-Ready Reports and Visuals
You receive a comprehensive deliverable package including:
- Annotated gene/miRNA tables
- Volcano plots, heatmaps, network diagrams
- Pathway and functional enrichment visuals
- Methods summary ready for supplementary materials
Why CD Genomics?
- Specialized expertise in extracellular RNA and vesicle-based sequencing
- High sensitivity workflows for low-input, fragmented RNA
- Transparent data delivery with no vendor lock-in
- Support for custom bioinformatics pipelines
To streamline your project from start to finish, our team remains available for post-delivery consultations and additional data interpretation support.
Conclusion – Get Reliable Results from Start to Finish
Successful Exosome RNA-Seq starts long before the sequencer runs—and extends far beyond raw FASTQ files. Each stage of the workflow, from thoughtful sample selection and vesicle isolation to tailored library construction and downstream bioinformatics, plays a decisive role in data quality and interpretability.
Researchers working with exosomal RNA must contend with low input amounts, fragmentation, and contamination risks. However, with the right technical strategy—and a partner experienced in extracellular RNA workflows—you can achieve meaningful, reproducible insights from these challenging but information-rich molecules.
At CD Genomics, we combine domain-specific expertise, flexible service options, and transparent data delivery to support your non-clinical exosome RNA research—whether your goal is biomarker discovery, mechanism exploration, or transcriptomic profiling.
Recommended Next Steps:
Download the Sample Submission Guidelines (PDF)
Ensure your RNA meets purity and input criteria.
Read: How to Prepare Samples for Exosomal RNA-Seq
Reduce processing errors and improve QC success.
Explore: Interpreting Exosomal RNA Sequencing Data
Learn how to convert expression matrices into biological insight.

 Sample Submission Guidelines
Sample Submission Guidelines


