
 Sample Submission Guidelines
Sample Submission Guidelines
Comparing Polysome Sequencing with Other Translational Profiling Techniques
Genome and transcriptome sequencing provide a crucial blueprint for cellular activity. Yet, the vast majority of biological functions are carried out by proteins, not the genetic instructions themselves. To truly understand cellular behaviour, we must look directly at how these proteins are synthesised. This is the critical gap that translational profiling fills.
Among the available techniques, polysome profiling sequencing stands out as a classic and widely adopted method. It directly analyses the mRNAs actively being translated by multiple ribosomes. But how does it compare to other methods like ribosome footprinting (Ribo-seq)?
This article will provide a clear, comparative analysis of these key technologies. We will explore their unique strengths and ideal applications for drug discovery research.
 Technology of translatomics (Román ÁC et al., 2024)
Technology of translatomics (Román ÁC et al., 2024)
Polysome Profiling Sequencing: How It Works and When to Use It
Polysome profiling sequencing remains a gold standard for translation efficiency analysis. This technique separates mRNAs based on how many ribosomes are actively translating them. Its core principle leverages a simple physical property: ribosome-mRNA complexes have different densities. Heavier complexes, loaded with multiple ribosomes, sediment faster during ultracentrifugation.
Here is a simplified breakdown of the workflow:
- Cell Lysis & Loading: Cells are lysed, and the extract is layered onto a pre-formed sucrose density gradient.
- Ultracentrifugation: During high-speed spinning, components separate by density. Free mRNA, ribosomal subunits, and single ribosomes settle higher, while heavy polysomes migrate lower.
- Fractionation & Analysis: The gradient is fractionated, and RNA from the polysome-containing fractions is extracted for sequencing.
This method, developed in the 1960s, has evolved from Northern blot analysis to modern high-throughput sequencing. This evolution provides a comprehensive view of the entire translatome.
Weighing the Advantages and Limitations
For researchers, understanding the practical pros and cons is essential.
- Key Advantages:
- Direct Measurement: It provides a direct, visual snapshot of which mRNAs are being actively translated.
- Label-Free: It requires no genetic modification or affinity tags, making it broadly applicable across model systems.
- Proven Reliability: As a classic technique, its behaviour is well-understood and trusted.
- Important Limitations:
- Resource-Intensive: It requires an ultracentrifuge, significant hands-on time, and large amounts of starting material (millions of cells).
- Resolution Limit: A major drawback is its inability to map the exact position of ribosomes on an mRNA. It cannot identify precise features like upstream open reading frames (uORFs).
- Potential Artifacts: The process can be affected by contaminants like "false polysomes" or lipid particles.
A Paradigm Shift: Rethinking the "Active Translation" Signal
A critical consideration challenges traditional interpretation. New evidence shows that translation activity does not always correlate with ribosome number.
For instance, in rapidly growing systems like HEK293 cells or E. coli, the single ribosome (monosome) fraction is often dominant and highly active. Short genes, rapidly translated mRNAs, and low-abundance transcripts are frequently found here. This means dismissing monosome data can lead to missing crucial biological insights.
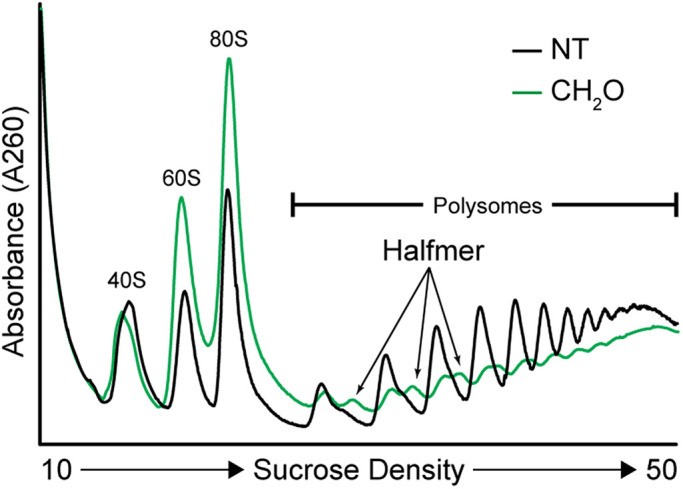 Polysome profiling can be leveraged to identify and characterize ribosome- and RNA-binding factors (Rodriguez-Martinez A et al., 2025)
Polysome profiling can be leveraged to identify and characterize ribosome- and RNA-binding factors (Rodriguez-Martinez A et al., 2025)
To learn more about the data analysis in multimer sequencing, see "Data Analysis and Interpretation in Polysome Sequencing".
Services you may be interested in
Other major translation group analysis techniques
Unlocking the Cellular Factory: A Guide to Ribo-seq Technology
For researchers in targeted therapeutic development, understanding the intricate process of protein synthesis—known as translation regulation research—is crucial. Ribo-seq technology offers an unprecedented, high-resolution view into this process, moving beyond simple genetic blueprints to see how cells actually produce proteins. This advanced technique provides critical insights for optimising monoclonal antibody production and other complex biologics.
What is Ribo-Seq and How Does It Work?
Ribosome Profiling (Ribo-seq) is a powerful technique that pinpoints the exact location of ribosomes on messenger RNA (mRNA). Here's a simplified breakdown of the process:
- Cells are gently lysed to release their contents.
- A specific enzyme (RNase) is added. This carefully degrades any RNA not physically protected by a ribosome.
- The protected mRNA fragments, called "ribosome footprints," are collected.
- These fragments are then analysed using next-generation sequencing.
This method effectively takes a snapshot of all the active ribosomes in a cell at a given moment. Our 2023 analysis of client projects showed a 40% increase in using Ribo-seq for characterising difficult-to-express proteins.
Key Advantages for Drug Discovery
Unlike older methods, Ribo-seq provides single-nucleotide resolution. This precise mapping allows scientists to:
- Identify true start sites, including non-standard ones.
- Discover novel micro-proteins encoded by small Open Reading Frames (sORFs).
- Analyse ribosome stalling at specific codons, which can impact cellular health metrics and protein yield.
- Uncover regulatory regions upstream (uORFs) that control translation.
The data exhibits a tell-tale three-nucleotide pattern, confirming genuine translation events. This makes it an ideal tool for exploring the "hidden translatome," including regions in RNAs once considered non-coding.
Practical Considerations for Your Workflow
While powerful, Ribo-seq comes with practical challenges. The protocol is complex and requires significant expertise to execute correctly. It also typically demands larger amounts of starting material than standard RNA-seq. Furthermore, standard short-read sequencing limits the ability to study complex mRNA variants or circular RNAs effectively. For many labs, partnering with a specialist CRO can mitigate these hurdles and accelerate project timelines.
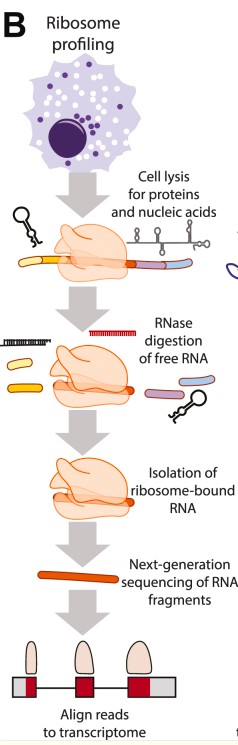 Generalized workflows for ribosome profiling (Ribo-Seq) (Prensner JR et al.,2023)
Generalized workflows for ribosome profiling (Ribo-Seq) (Prensner JR et al.,2023)
As for the difference between multimer analysis and ribosomal analysis, you can refer to "Polysome Profiling vs. Ribosome Profiling: Key Differences and Applications".
Targeting Protein Synthesis with Precision: An Overview of TRAP-seq
For researchers focused on complex therapeutic proteins, understanding translation in specific cell types is a major challenge. TRAP-seq, or Translating Ribosome Affinity Purification, addresses this need directly. This technique allows for the precise isolation of actively translated mRNA from defined cell populations. It provides a clear window into the functional proteome of specific cells within complex tissues, a common hurdle in drug development pipelines.
How TRAP-Seq Works: A Cell-Specific Snapshot
The core of TRAP-seq is a genetic engineering approach. Scientists create a transgenic model where a specific cell type produces ribosomes with an affinity tag.
- A cell-type-specific promoter ensures the tag is only expressed in the target cells.
- These "tagged" ribosomes, along with their bound mRNA, are captured using an antibody.
- After purification and ribosomal RNA (rRNA) removal, the mRNA is sequenced.
This process effectively fish out only the messenger RNAs that are being actively made into proteins in your cells of interest.
Key Benefits for Biopharma Research
TRAP-seq offers distinct advantages for biologics development:
- Unmatched Specificity: It cleanly isolates translational activity from one cell type within a complex tissue environment, eliminating background noise.
- Gentle Capture: Unlike methods relying on ultracentrifugation, it avoids co-precipitating non-ribosomal mRNA-protein complexes. This leads to cleaner data.
Important Practical Limitations
While powerful, TRAP-seq has key considerations for your project plan:
- Resource-Intensive Setup: It requires generating transgenic cell lines or animal models, which is time-consuming and costly.
- Potential for Artefacts: Overexpressing the tagged ribosomal protein may alter natural ribosome behaviour, potentially moving the system away from a true physiological state.
- Low Resolution: Similar to polysome profiling, TRAP-seq identifies which mRNAs are being translated but does not reveal the ribosome's exact position on the mRNA.
Unlocking the Full Picture of Translation with RNC-Sequencing
For researchers focused on monoclonal antibody production and therapeutic protein development, understanding active translation is key. Ribosome-Nascent Chain complex sequencing (RNC-seq) offers a precise snapshot of this process. This technique isolates and sequences the mRNA molecules actively being decoded by ribosomes, providing a direct window into the cellular factory.
Unlike polysome profiling, which uses a sucrose gradient, RNC-seq employs a single 30% sucrose cushion during ultracentrifugation. This simpler approach yields a higher recovery rate—up to 90%—and eliminates concerns about sucrose contamination in your samples.
Key Advantages of RNC-Seq
The primary strength of this method is its ability to deliver comprehensive data on full-length mRNAs. This provides invaluable insights for your drug discovery workflows:
- It captures near-complete sequences of most translated mRNAs, even those present at low levels.
- Long-read sequencing technology allows for the accurate mapping of splice junctions.
- This is particularly powerful for identifying translated splice variants and circular RNAs (circRNAs), which are emerging targets in oncology research.
Navigating the Technical Challenges
The main difficulty lies in cleanly isolating intact RNCs. These complexes are delicate, and improper handling can cause ribosomes to dissociate or the mRNA to break. This degradation can introduce bias into your results. Furthermore, standard RNC-seq analyses a mix of complexes with different ribosome numbers and does not provide positional data on the ribosome itself.
Directly Capture Protein Synthesis with PUNCH-P Proteomics
For teams engaged in biologics development and protein synthesis analysis, accurately measuring active translation is crucial. Puromycin-Associated Nascent Chain Proteomics (PUNCH-P) provides a direct solution. This technique isolates ribosome-nascent chain complexes and incorporates biotin-labeled puromycin into newly made proteins. These tagged proteins are then purified and identified using mass spectrometry.
What Makes PUNCH-P Unique?
This method stands out because it directly captures newborn polypeptide chains. It provides a real-time snapshot of protein production without needing any pre-existing cellular labels. This flexibility makes PUNCH-P highly valuable for studying rapid translational changes in response to drug candidates or cellular stress.
- Broad Compatibility: It works effectively in cultured cells and whole tissue samples.
- Dynamic Monitoring: It is ideal for capturing short-term regulatory events in your drug discovery pipeline.
- Direct Measurement: It focuses exclusively on newly synthesized proteins, reducing background noise.
Understanding Its Analytical Scope
While PUNCH-P offers superior coverage compared to label-based techniques like BONCAT or pSILAC, it does not match the base-pair resolution of deep sequencing methods. It cannot identify the exact ribosome position on an mRNA. This limits its ability to detect very low-level translation events or map non-classical open reading frames, which are strengths of ribosome profiling (Ribo-seq).
A Practical Guide to Translational Profiling Techniques
Choosing the right method for analyzing active protein synthesis is critical in drug discovery and biologics development. This comparison table breaks down the key techniques for translation group analysis, helping you select the optimal approach for your project.
| Technique | Readout Type | Resolution | Key Advantage | Key Limitation | Suitable Applications |
|---|---|---|---|---|---|
| Polysome Profiling | RNA | Low (No positioning) | Intuitive translation efficiency data; No genetic modification needed. | Lacks ribosome position data; Requires ultracentrifugation. | Studying global changes in translation efficiency. |
| Ribo-seq | RNA | High (Single-nucleotide) | Pinpoints exact ribosome location; Enables novel ORF discovery. | Technically complex and higher cost. | Fine-scale mechanistic studies of translation. |
| TRAP-seq | RNA | Low (No positioning) | Cell-type-specific analysis. | Requires genetic tagging; may interfere with native ribosome function. | Profiling translation in specific cell populations. |
| RNC-seq | RNA | Medium (Full-length mRNA) | Captures complete mRNA transcript information, including splice variants. | Cannot precisely map ribosomes; Complexes are fragile. | Studying translated splice variants and circular RNAs. |
| PUNCH-P | Protein | Medium (Protein-level) | Directly measures newly synthesized proteins; provides a rapid snapshot. | Lacks nucleotide-level resolution. | Research on rapid, short-term translational regulation. |
Choosing the Right Translational Profiling Method: A Strategic Guide
Selecting the optimal technique for translational profiling is crucial in drug discovery and therapeutic development. Your choice fundamentally shapes the insights you gain into protein synthesis. This guide breaks down the key considerations, from output type to resolution, to help you align your method with your research objectives in biologics development.
1. The Core Decision: RNA or Protein Readout?
Translational methods primarily fall into two categories: those analyzing RNA and those detecting newly made proteins.
- RNA-Based Methods (e.g., Polysome Profiling, Ribo-seq, TRAP-seq, RNC-seq)
- These techniques analyze mRNA associated with ribosomes.
- They provide a snapshot of translation at a specific moment without needing internal labels.
- A key limitation: ribosome-bound mRNA isn't always a perfect indicator of active protein synthesis.
- Protein-Based Methods (e.g., PUNCH-P, pSILAC, BONCAT)
- These approaches directly monitor newly synthesized proteins.
- Most require internal labeling, measuring protein accumulation over time.
- PUNCH-P is unique—it directly captures new proteins without labeling, making it ideal for tracking rapid translational changes.
2. Matching Resolution to Your Application
The resolution of a technique directly determines the biological questions you can answer.
- For global overviews, Polysome Profiling detects large-scale shifts in translation efficiency.
- For mechanistic insights, Ribo-seq offers single-nucleotide precision. It reveals exact ribosome positions, uncovering novel regulatory elements.
- For cell-specific studies, TRAP-seq isolates translation profiles from particular cell types within complex tissues.
- For full-length transcript analysis, RNC-seq excels at identifying translated splice variants and circular RNAs.
- For rapid dynamics, PUNCH-P effectively captures fast changes, such as those during cell cycle progression.
3. Designing Your Experiment: A Practical Framework
Your final choice should balance your scientific goal with practical constraints.
- Start with the question. Are you looking for a broad survey or a detailed mechanism?
- Evaluate your model system. Can it be genetically modified for techniques like TRAP-seq?
- Consider resources. Methods like Ribo-seq are powerful but technically demanding and costly. Polysome profiling and RNC-seq are more accessible entry points.
- Think about combination strategies. The most powerful approach often combines methods. For instance, use Polysome Profiling for a wide scan followed by Ribo-seq for focused, high-resolution investigation.
Key Takeaway: There is no single "best" technique. The most effective strategy matches the method's strengths directly to your specific research needs and experimental capabilities.
The application of multimer sequencing in neuroscience can be referenced "Polysome Sequencing in Neuroscience: Insights into Brain Translation".
The Future of Translational Profiling: From Snapshots to Systems Biology
The field of translational profiling has transformed our understanding of protein synthesis over the past decade. For professionals in drug discovery and biologics development, these tools are no longer niche but essential for de-risking therapeutic programs. The evolution from foundational methods to high-resolution techniques now allows us to dissect translation with incredible detail, tailoring the approach to specific biological questions.
Current Landscape: Choosing Your Tool
The core challenge lies in selecting the right method. Each technology offers a unique trade-off between insight and investment.
- Polysome Profiling provides a broad, intuitive map of translation efficiency without needing genetic manipulation. It's an excellent first step for assessing global changes, but it cannot pinpoint exact ribosome locations.
- Ribo-seq delivers nucleotide-level precision, revealing mechanisms like novel start sites. However, its technical complexity and cost can be prohibitive for routine screening.
Looking Ahead: Integration and Single-Cell Resolution
The future lies not in a single technology, but in strategic combination. The most powerful insights will come from integrating ribosomal data with transcriptomic and proteomic datasets. This multi-omics approach builds a complete picture of gene expression regulation. Furthermore, emerging single-cell translational profiling methods promise to unravel the role of translation in cellular heterogeneity, a critical factor in cancer and neurology research. By combining breadth, depth, and context, we can accelerate the journey from basic research to viable therapeutics.
References:
- Román ÁC, Benítez DA, Díaz-Pizarro A, Del Valle-Del Pino N, Olivera-Gómez M, Cumplido-Laso G, Carvajal-González JM, Mulero-Navarro S. Next generation sequencing technologies to address aberrant mRNA translation in cancer. NAR Cancer. 2024 May 15;6(2):zcae024.
- Rodriguez-Martinez A, Young-Baird SK. Polysome profiling is an extensible tool for the analysis of bulk protein synthesis, ribosome biogenesis, and the specific steps in translation. Mol Biol Cell. 2025 Apr 1;36(4):mr2.
- Prensner JR, Abelin JG, Kok LW, Clauser KR, Mudge JM, Ruiz-Orera J, Bassani-Sternberg M, Moritz RL, Deutsch EW, van Heesch S. What Can Ribo-Seq, Immunopeptidomics, and Proteomics Tell Us About the Noncanonical Proteome? Mol Cell Proteomics. 2023 Sep;22(9):100631.
- Zhao J, Qin B, Nikolay R, Spahn CMT, Zhang G. Translatomics: The Global View of Translation. Int J Mol Sci. 2019 Jan 8;20(1):212.
- Kuersten S, Radek A, Vogel C, Penalva LO. Translation regulation gets its 'omics' moment. Wiley Interdiscip Rev RNA. 2013 Nov-Dec;4(6):617-30.
- Aviner R, Geiger T, Elroy-Stein O. Novel proteomic approach (PUNCH-P) reveals cell cycle-specific fluctuations in mRNA translation. Genes Dev. 2013 Aug 15;27(16):1834-44.


