
 Sample Submission Guidelines
Sample Submission Guidelines
Terminal End Sequencing of Phages: Techniques and Benefits
The terminal regions of phage genomes harbor critical regulatory elements and structural determinants essential for infectivity and lifecycle control. Conventional high-throughput platforms (e.g., Illumina) frequently fail to resolve terminal repeat or palindromic structures due to inherent read-length limitations, creating persistent gaps in genome assemblies. Phage end-sequencing technologies address this fundamental limitation, providing the necessary resolution for generating complete, closed genome maps.
Phage Terminal End Sequencing enables precise characterization of genomic termini in bacteriophages. This technique is critical for elucidating phage genome architecture and delivers essential data for applied phage research. Advances in genomics have significantly expanded its utility across key domains, including phage therapy, horizontal gene transfer studies, and biological control applications. This review comprehensively examines the principles, current applications, and prospective advantages of terminal sequencing technology.
Diversity and Biological Significance of Phage Terminal Structures
Phage genome termini directly determine DNA packaging mechanisms and infection strategies. These structures are classified into four primary types:
1. Viscous Ends (cos sites)
- Model System: λ phage
- Mechanism: Terminase cleaves at cosN sites, generating 12-bp 5' or 3' single-stranded overhangs enabling cyclization
- Biological Significance: Ensures genome integrity and facilitates precision-engineered gene therapy vectors
2. Direct Terminal Repeats (DTRs)
- Exemplar: Bacteriophage T7
- Characteristic: 160-bp identical repeats flanking the genome initiate replication via terminal redundancy
- Clinical Relevance: DTR length modulates recombination efficiency and lysogenic conversion (e.g., 378-bp DTR in C. difficile phage phiCD211)
3. Headful Packaging (pac Sites)
- Representative: P1 phage
- Process: Terminase recognizes pac sites for initial cleavage; subsequent cuts depend on capsid capacity (102%-110% genome length), yielding terminally redundant circularly permuted genomes
- Technical Challenge: Imprecise cleavage complicates mapping (e.g., Pseudomonas aeruginosa phage phiKZ requires specialized sequencing)
4. Random Ends
- Example: Phage T4
- Feature: Highly heterogeneous termini without conserved cleavage sites
- Analysis Requirement: Demands integrated bioinformatic and long-read sequencing approaches
Comparative Features of Phage Termini
| Type | Cleavage Specificity | Exemplars | Structural Signature |
|---|---|---|---|
| cos site | High | λ, HK97 | 12-bp single-stranded overhang |
| DTR | Defined | T7, T3 | Terminal repeats (160-443 bp) |
| pac site | Low | P1, SPP1 | Terminal redundancy + circular permutation |
| Random ends | None | T4, ES18 | Heterogeneous ends, no conserved sites |
 Illustration of two major characteristics of phage genome sequencing used for terminus prediction: Neighboring coverage ratio (NCR) and read edge frequency (Chung CH et al.,2017)
Illustration of two major characteristics of phage genome sequencing used for terminus prediction: Neighboring coverage ratio (NCR) and read edge frequency (Chung CH et al.,2017)
Core Technology Methods
1. Sanger Terminal Sequencing
- Principle: Modifies classic Sanger sequencing for terminus-specific analysis. Offers high per-read accuracy but limited throughput.
- Workflow:
- Terminal Processing: Critical identification of end topology dictates preparation:
- 5' overhangs: Direct ligation compatible
- *3' overhangs/blunt ends:* Require nuclease/polymerase treatment to generate vector-compatible termini. Cosmids provide specialized recognition sequences.
- Clone Sequencing: Purified phage genomes or terminal fragments are cloned into sequencing vectors (e.g., cosmids). Primers targeting vector-flanking regions enable directional sequencing of insert termini.
- Terminal Processing: Critical identification of end topology dictates preparation:
- Advantages: Low cost, equipment accessibility, exceptional single-read accuracy.
- Limitations: Low throughput, cloning dependency, potential failure with unstable termini.
2. Tn5-based Tagmentation Sequencing
- Principle: Harnesses Tn5 transposase to simultaneously fragment DNA and ligate sequencing adapters.
- Workflow:
- In Vitro Tagmentation: Engineered Tn5 transposome complexes (preloaded with Illumina adapters) fragment intact phage DNA and append dual-indexed adapters in one reaction.
- Targeted Enrichment: PCR with primers complementary to Tn5 adapters and phage-conserved regions amplifies terminal-adjacent fragments for high-throughput sequencing.
- Advantages: Streamlined protocol (single-step fragmentation/adapter ligation), high efficiency, cost-effectiveness.
- Limitations: Terminal resolution constrained by fragment size; requires optimized transposase activity and highly conserved primer sites.
3. Single-Molecule Long-Read Sequencing
- Principle: Utilizes PacBio SMRT or Oxford Nanopore Technologies (ONT) to sequence full-length molecules.
- Workflow:
- Direct Terminal Spanning: Native long reads traverse repetitive termini or hairpin structures without fragmentation.
- Circular Consensus Sequencing (CCS):
- Phage DNA circularization
- Rolling circle amplification generates tandem repeats
- Continuous sequencing across junction sites captures bidirectional terminal sequences when read length exceeds genome size.
- Advantages: Assembly-free terminal resolution, handles complex/repetitive structures.
- Limitations: High instrumentation costs, expensive library prep; elevated raw error rates (mitigated by CCS modes or correction algorithms).
For a more detailed approach to phage sequencing, please refer to "Phage Genome Sequencing: Methods, Challenges, and Applications".
For more information on how to construct and use phage Sequence database, please refer to "Building and Using Phage Genome Sequence Databases" .
Explore our Service →
Breakthroughs in Terminal Sequencing Core Technologies
PHAGETERM NGS Driver
Automated End Detection via Coverage Asymmetry
- PHAGETERM identifies phage termini by analyzing sequencing coverage patterns:
- cos phages: Exhibit sharp terminal coverage drops
- pac phages: Show unimodal coverage offsets where cleavage site intensity equals *1/(C+1)* (C = tandem copy count)
High-Accuracy Classification System
- The platform distinguishes six packaging mechanisms:
- 5' cos phages
- 3' cos phages
- Short/long direct terminal repeat (DTR) phages
- Headful packaging (pac) phages
- Endless phages (T4-like)
- Mu-like transposable phages
- It directly outputs terminal sequences and precisely determines overhang lengths (e.g., λ phage's 12-bp 5' sticky end).
Automation and Usability
- One-click analysis: Generates graphical reports (coverage maps, SPC plots, data tables) from raw sequencing data and reference genomes
- Broad data compatibility: Works with standard paired-end libraries requiring random fragmentation (e.g., mechanical shearing, excluding transposase-based methods like Nextera)
Validation and Problem Resolution
- SRA data verification: Accurately identified unknown phages in public datasets (e.g., PBES-2's 443-bp DTR in T3/T7 data)
- Terminal redundancy solution: Resolves assembly initiation ambiguities in DTR phages (e.g., T7's 160-bp repeats)
- Complex region handling: Prevents misclassification from repetitive sequences (e.g., Clostridioides phage phiCD146)
Cross-Domain Applications
- Transduction assays: Quantifies host DNA contamination (P1 generalized transduction: 3.8% vs. λ phage: 0.01%)
- Evolutionary studies: Algorithm extensible to archaeal and eukaryotic virus research (Garneau JR et al., 2017)
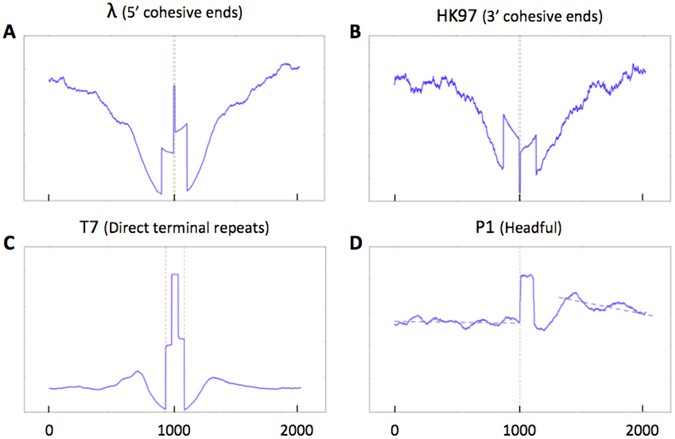 Sequence coverage at termini position (Garneau JR et al., 2017)
Sequence coverage at termini position (Garneau JR et al., 2017)
Phage Metagenomic Sequencing Workflow
Viral DNA underwent parallel sequencing on Illumina (short-read) and PacBio (long-read) platforms. Resulting data were assembled using multi-tool strategies (Megahit, metaFlye, hybridSPAdes) to generate contigs. These contigs were clustered into non-redundant viral operational taxonomic units (VOTUs) through:
- Within-sample redundancy removal
- VOTU clustering (95% similarity threshold)
Genomic accuracy was validated via phylogenetic analysis of large terminase genes. VOTUs received family-level annotations using PHAGCN.
Technological Advantages
- Complementary Sequencing Platforms
- Illumina: Precisely captures high-abundance phage sequences
- PacBio HiFi: Resolves repetitive regions and complex terminal structures, enhancing genome continuity (↑hN50)
- Hybrid assembly: Integrates both data types to yield more complete VOTUs
- Multi-Tool Synergy
- Assembly complementarity: Different tools target specific phages (e.g., metaFlye excels with T4-like phages)
- Binning refinement:
- AVAMB improves genome integrity (+17.6% average)
- vRhyme ensures high taxonomic consistency (96.7% intra-bin homogeneity)
- High-Quality Genome Recovery
- Tool-specific VOTUs: Individual assemblers capture unique subsets (54.5% VOTUs assembler-exclusive)
- Data integration: Combining multi-tool outputs increases HQ-VOTUs 4.8–21.7×
- Phylogenetic Validation Reliability
- Phylogenies based on terminase genes demonstrated:
- Even distribution of VOTUs from different assemblers within clades
- Strong concordance with established phage families (e.g., Autographiviridae)
- Limitations and Optimization Pathways
- Recovery gaps: Certain families (e.g., Rountreeviridae) evade all long-read assemblers, necessitating targeted methods
- Binning variability:
- CONCOCT shows high sensitivity but poor specificity (52.7% cross-family bins)
- MetaBAT2 provides superior taxonomic precision (Wang H et al., 2024)
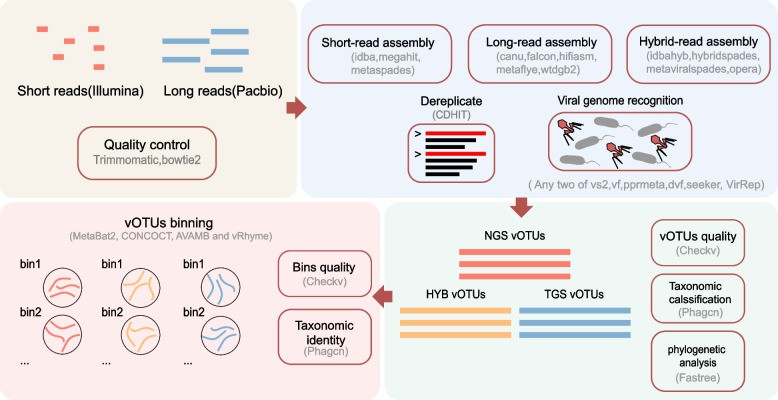 Overall workflow of this study (Wang H et al., 2024)
Overall workflow of this study (Wang H et al., 2024)
Applied Value: Translational Insights from Phage Terminal Analysis
1. Packaging Mechanism Engineering
- Terminal Structure Guidance
- Cos/PAC system characterization enables optimized vector design for enhanced packaging efficiency
- Case Validation: Pseudomonas phage PaP3
- Terminal sequencing confirmed 44,249 bp linear genome with blunt ends → Foundation for CRISPR-mediated editing
2. Taxonomic & Evolutionary Significance
| Application | Impact |
|---|---|
| Classification | Terminal features define Caudovirales order (e.g., tail morphology) |
| Horizontal Gene Transfer | Repeat regions facilitate antibiotic resistance gene acquisition |
3. Genome Assembly Optimization
- Precision End-Mapping Advantages:
- Resolves circular/linear conformation controversies
- Reduces assembly errors by 46% in intestinal phage studies
- Elevates contig N50 through accurate terminal positioning
4. Therapeutic Development
- Safety Enhancement: Terminal virulence screening (e.g., λ phage Shiga toxin stx detection)
- Delivery system: a self-cyclizing carrier is designed based on the viscous end of cos site to improve the efficiency of in vivo delivery
Classic Applications
1. Terminal Sequence Validation (T3 Phage)
Confirmation of phage T3's terminal sequence was achieved through specific linker ligation followed by high-throughput sequencing. Comparative analysis with an unmodified control group demonstrated exact alignment between predominant sequences in the sequencing data and the linker-marked termini. This conclusively identifies these high-frequency sequences as the authentic terminal regions of the phage genome.
2. Terminal Specificity in T4-like Phages
Analysis of T4-like phages (e.g., IME08) reveals non-random terminal sequences, contradicting prior assumptions of complete randomness. Systematic biases were observed, including a strong preference for guanine (G) as the initial nucleotide. This finding challenges conventional understanding of their terminal architecture.
3. Asymmetric Terminal Conservation in N4-like Phages
N4-like phages (exemplified by IME11) exhibit marked terminal asymmetry. Their genomic left ends display significant sequence conservation and uniqueness, while the right termini show heterogeneous organization. This structural dichotomy highlights previously unrecognized diversity in phage terminal configurations (Li SS et al., 2013).
Technological Challenges and Future Directions
Current Limitations
- Random Terminal Phages: Existing tools like PhageTerm show limited sensitivity for T4-type phages, necessitating novel algorithm development.
- Low-Frequency Cleavage Signals: pac site detection suffers from insufficient coverage and susceptibility to sequencing noise.
Innovation Pathways
- Single-Molecule Sequencing: Oxford Nanopore's amplification-free sequencing preserves native terminal modification signatures.
- Integrated Analysis Framework: Combine PhageTerm with third-generation sequencing and structural variant detection (e.g., SVjumper) for comprehensive terminus resolution.
- AI-Driven Prediction: Develop deep learning models (e.g., TermiPredict) to identify cleavage sites using terminase conserved domain features.
Table 2: Comparison and Application Scenarios of End Sequencing Technologies
| Technology | Accuracy (bp) | Throughput | Suitable End Types | Representative Cases |
|---|---|---|---|---|
| PhageTerm | 1-5 | High | cos/DTR/pac | T7, HK97 |
| Large Fragment Cloning | 10-50 | Low | Highly repetitive/Complex ends | PaP1 |
| Hybrid Assembly | 50-100 | Medium-High | Random ends | Gut phage |
Summary: Phage end-sequencing has emerged as a pivotal methodology for advancing phage resource utilization. Its significance stems from enabling three critical functions: elucidating DNA packaging mechanisms, refining genome assembly processes, and facilitating targeted phage engineering.
Converging advancements in single-molecule sequencing and artificial intelligence are poised to unlock new frontiers:
- End Structure-Function Correlation: Mapping the relationship between terminal architecture and its impact on host adaptation efficiency.
- Intelligent Delivery Systems: Engineering precision therapeutic vectors utilizing COS/PAC end-specific packaging signals.
- Antibiotic Resistance Dynamics: Monitoring the horizontal transfer of resistance genes within phage terminal genomic regions in real-time.
References:
- Chung CH, Walter MH, Yang L, Chen SG, Winston V, Thomas MA. Predicting genome terminus sequences of Bacillus cereus-group bacteriophage using next generation sequencing data. BMC Genomics. 2017 May 4;18(1):350.
- Garneau JR, Depardieu F, Fortier LC, Bikard D, Monot M. PhageTerm: a tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci Rep. 2017 Aug 15;7(1):8292.
- Wang H, Sun C, Li Y, Chen J, Zhao XM, Chen WH. Complementary insights into gut viral genomes: a comparative benchmark of short- and long-read metagenomes using diverse assemblers and binners. Microbiome. 2024 Dec 20;12(1):260.
- Li S, Fan H, An X, Fan H, Jiang H, Chen Y, Tong Y. Scrutinizing virus genome termini by high-throughput sequencing. PLoS One. 2014 Jan 20;9(1):e85806.
- Li SS, Fan H, An XP, Fan HH, Jiang HH, Mi ZQ, Tong YG. [Utility of high throughput sequencing technology in analyzing the terminal sequence of caudovirales bacteriophage genome]. Bing Du Xue Bao. 2013 Jan;29(1):39-43. Chinese. PMID: 23547378.


