
 Sample Submission Guidelines
Sample Submission Guidelines
Building and Using Phage Genome Sequence Databases
As Earth's most abundant biological entities, bacteriophages (viruses infecting bacteria) play pivotal roles in microbial ecology, pathogenesis research, and therapeutic applications. High-throughput Phage Genome Sequencing has rapidly generated vast datasets of phage genetic material. To make sense of this wealth of information, sequence databases—such as PhageScope and PhagesDB—are indispensable for storing, annotating, and visualizing genomic data. Conversely, as these databases continue to integrate new high-quality sequences, they further enhance the power and precision of sequencing-based analyses, forming a virtuous cycle that underpins modern phage genomics.
 Overview of PhageScope database (Wang RH wt al., 2024)
Overview of PhageScope database (Wang RH wt al., 2024)
Constructing the Bacteriophage Genome Database
1. The Core Challenge: Why We need tough standards
Imagine trying to assemble a global library where every book is in a different language and format. This was the state of phage data. Building a reliable database requires a rigorous, multi-layered approach to standardize information from diverse sources:
2. Diverse Data Acquisition Sources
| Source Type | Key Specifications |
|---|---|
| Public Database Integration | NCBI GenBank/RefSeq/ENA sources; requires phage-specific filtering |
| Metagenomic Studies | Reconstructing viral genomes from complex environmental or host tissue samples. |
| Laboratory Isolates | Cultured phage genomes with experimental validation |
| Draft Genome Submissions | Temporarily hosted incomplete sequences with completion requirements |
2. Source-Specific Processing Standards
Public Databases
- Screening: Automated taxonomic filters extract phage entries
- Compliance Tiering:
- Complete: Circularized + annotated
- High-Quality Draft: N50 > 50 kb
- Draft: N50 > 10 kb
- Fragment: Unassembled segments
- Deduplication: CD-HIT-EST clustering (99% identity threshold)
Metagenomic vMAGs
- Identification Pipeline:
- VirSorter2 (marker-based)
- DeepVirFinder (k-mer/AI prediction)
- vRhyme (cross-sample binning)
- QC Thresholds: CheckV-verified ≥50% completeness + ≤10% contamination
Laboratory Isolates
- Submission Requirements:
- Host strain documentation (e.g., ATCC no.)
- EM structural verification
- Experimental validation (plaques/growth curves)
- Priority Handling: Expedited review and featured display
Draft Submissions
- Provisional Status: Temporary ID with 1-year completion deadline
- Assembly Incentives: Preference for hybrid Nanopore+Illumina submissions
- Non-Compliance: Automatic downgrade to "Deprecated" status
Each source undergoes specific processing and strict quality checks to ensure it meets a defined grade—from "Complete" to "Draft"—before entering the database.
The Engine Room: How Data is Processed and Annotated
Once collected, the data must be cleaned, standardized, and interpreted.
1. Rigorous Quality Control:
Every genome is put through a multi-step validation process to check its completeness and remove any contaminating DNA from the host bacteria or other organisms.
- Assessment Tools:
- CheckV: Quantifies completeness (%) and contamination (%)
- BUSCO (viral gene set): Evaluates core gene preservation
- Certification Criteria for "Complete" Status:
- Terminal overlap ≥10 bp
- ≥90% CheckV completeness
- Presence of ≥4 core viral genes (e.g., terminase large subunit, capsid protein)
Note: Draft/MAG entries must display integrity/contamination metrics prominently
2. Standardized "Labeling":
Each sequence is tagged with consistent, rich metadata—like the host bacterium, GPS coordinates of where it was found, and sampling date. This turns a raw sequence into a meaningful biological story.
- FASTA Header Requirements:
- >DatabaseID|Host_Genus|IsolationSource|Date[YYYY-MM-DD]
- Example: >PhageDB_KT003|Pseudomonas|Marine_sediment|2023-05-17
3. Minimum Metadata Standards
| Field Type | Requirements |
|---|---|
| Mandatory | Host taxonomy (genus-level), GPS coordinates, Sampling date |
| Recommended | Environmental parameters (pH/temperature), ≥50× sequencing depth, Assembly tools (e.g., SPAdes v3.15.5) |
4. Contamination Control Protocol
- Three-Stage Filtration:
- Primary Screening: Remove host-derived sequences (e.g., 16S rRNA) via NT database alignment
- Deep Classification: Discard contigs with >5% non-viral matches using Centrifuge
- Targeted Purification: Eliminate residual host fragments with k-mer databases (HostCleanse)
Comparison of Mainstream Phage Databases
| Database Name | Core Features | Application Scenarios |
|---|---|---|
| PhageScope | Integrates 15 analytical tools, supports automated annotation, comparative genomics, and visualization (e.g., circular genome maps) | Virulence factor mining, phage therapy target screening |
| SEA-PHAGES/PhagesDB | Focuses on actinophages, combined with educational programs; pdm_utils toolkit supports MySQL database management for dynamic updates | Teaching and research, genome annotation iteration |
| MGV | Human gut virome database containing 189,680 viral genomes covering 54,118 vOTUs with 81% host association accuracy | Study of gut microbiota-phage interactions |
| GPD | Catalogs 142,809 non-redundant gut phage genomes and discovered the novel taxon "Gubaphage" | Metagenomic analysis of diseases (e.g., inflammatory bowel disease) |
From raw data to trusted information: a brief description of quality control and standardization pipelines.
1. Core Functional Element Prediction
- tRNA Identification: tRNAscan-SE v2.0 (-B -O parameters)
- rRNA Detection: Barrnap v0.9 (virus mode: --vir)
- Non-coding RNA: Infernal + Rfam 14.0
- ORF Prediction:
- Prodigal v2.6 (prokaryotic mode: -p meta)
- MetaGeneMark v4.0 (cross-validated)
- Retention threshold: CDS ≥30 amino acids with valid start codon (ATG/GTG)
2. Functional Annotation Protocol
Tiered Assignment System
- Primary Annotation: Diamond BLASTP vs. PHROGS (e-value ≤1e⁻⁵, coverage ≥70%)
- Secondary Annotation: InterProScan domain analysis (Pfam/SUPERFAMILY)
- Tertiary Annotation:
- DeepFri structure-based GO term assignment
- Conflict resolution: PHROGs > InterPro > UniProt hierarchy
3. Integrated Genomic Feature Prediction
| Feature Type | Tool/Method | Key Specification |
|---|---|---|
| Lytic/Lysogenic Modules | Pharokka | Integrase + att site mapping |
| Promoters/Terminators | BPROM (σ70) + Arnold | Regulatory element screening |
| tRNA-Associated Genes | Genomic proximity analysis | ≤10 kb flanking regions |
4. CRISPR System Analysis
- Spacer Identification: CRISPRCasTyper v2.4.1
- Target Prediction: CRISPRTargetDB alignment
- Anti-CRISPR Genes: AcrFinder + custom HMM profiles
5. Quality Assurance Workflow
Workflow Implementation
- Automated Phase: Standardized processing via pipeline managers
- Manual Intervention: Required for evolutionarily significant features:
- First-reported phage-host pairs
- Unannotated gene clusters (≥3 consecutive unknown CDS)
- Novel anti-CRISPR or toxin candidates
Multi-Tiered Metadata Architecture for Phage Genomes
1. Core Metadata Specifications
- Storage Implementation:
- Embedded in sequence files: INSDC-compliant FASTA headers (## comment lines)
- Structured database storage:
| Table | Content |
|---|---|
| Source | Host organism, isolation source |
| Collection | GPS coordinates, date (YYYY-MM-DD) |
| Processing | Laboratory protocols, personnel |
| Publication | DOI/PubMed IDs, citation links |
2. Ecological & Phenotypic Extensions
Dynamic Field System (Optional but Recommended)
- Host Range: Comma-delimited verified hosts
- Example: Salmonella enterica, Escherichia coli
- Lytic Profile: Quantitative parameters
- Format: latent_period=20min; burst_size=150PFU
- Morphology: Dual-component documentation
- EM image repository links
- ICTV classification codes (e.g., Caudoviricetes; Siphoviridae)
- Stability Data: Temperature/pH tolerance ranges
3. Ontology-Driven Standardization
- Controlled Vocabulary Enforcement:
| Domain | Ontology | Implementation Example |
|---|---|---|
| Host Taxonomy | NCBI Taxonomy ID | taxon:562 → Escherichia coli |
| Environment | ENVO | ENVO:00010606 → Riverine water |
| Experimental Methods | OBI (Ontology for Biomedical Investigations) | OBI:0000070 → DNA extraction |
Database Architecture & Implementation Framework
1. Backend Storage System
- Hybrid Data Management:
| Database Type | Function | Optimization |
|---|---|---|
| PostgreSQL | Core metadata & annotations | Index-accelerated queries |
| MongoDB | Dynamic datasets (host ranges) | Flexible schema for lab data |
| Neo4j | Host-phage interaction networks | Graph relationship modeling |
2. Scalable File Management
- Sequence Storage: FASTA files in HDF5 block compression (70% space reduction)
- Search Optimization:
- Weekly automated BLAST index updates
- Distributed Diamond index sharding
3. Web Interface & API
- Advanced Search Capabilities:
- Sequence similarity: BLAST/BLAT + MASH prescreening
- Combinatorial queries (e.g., "Terminases AND marine hosts")
- Taxonomic tree filtering
4. Integrated Visualization Suite
| Tool | Functionality | Output Formats |
|---|---|---|
| JBrowse 2 | Gene/domain/variant visualization | Interactive SVG/HTML |
| PhyloViz | Host range heatmaps | Dynamic PDF reports |
| PPanGGOLiN | Comparative genomic island analysis | Vector graphics + HTML |
Summary of key innovation points
- Hierarchical management of dynamic data
- Automatic ranking based on data quality (Complete/Draft/Deprecated)
- Establish a "Gold Standard Dataset" for laboratory strains
- Guarantee of whole process reproducibility
- All annotation tool versions are solidified in the Docker container
- Provide workflow. YML for user local re-annotation
- Multi-dimensional relevance retrieval capability
- Support "Environment → host → phage → gene function" penetrating query
- Integrating phylogenetic trees with geographic information system data
- The "Dark matter" strategy
- Initiate automatic structure prediction (AlphaFold2) for unknown gene clusters
- Establishment of the "Phage Orphan Gene Repository"
For a more detailed approach to phage sequencing, please refer to "Phage Genome Sequencing: Methods, Challenges, and Applications".
To see how the Illumina platform can deep sequence phage libraries, see "Deep Sequencing of Phage Libraries Using Illumina Platforms".
Understanding the role of NGS data in quality control of phage display libraries can be referenced "Quality Control for Phage Display Libraries with NGS Data".
Powering Discovery: Key Applications for Researchers
- Precision Taxonomy & Phylogeny: Moving beyond morphology, databases use whole-genome analysis for accurate classification and large-scale evolutionary mapping.
- Novel Phage Discovery: They facilitate the discovery of new phages and unique genes, including those for host-range determination and novel anti-CRISPR (Acr) systems, directly feeding into therapeutic discovery programs.
- Phage Therapy Development: They serve as a primary screen for identifying pathogen-targeted phage candidates, predicting host range, and conducting critical safety assessments for lysogeny and virulence genes.
- Metagenomic Analysis: Acting as a essential reference, they enable researchers to identify and classify viral sequences within complex mixtures of DNA from the human gut, oceans, or soil.
Case in Point: Discovering Hidden Diversity
1. Taxonomy & Phylogeny
- Precision Classification
- Whole-genome similarity analysis using VIRIDIC (ICTV-standard metrics)
- Core gene phylogenies overcoming morphological limitations
- Evolutionary Mapping
- Large-scale phylogenetic reconstructions
- Divergence pattern analysis across phage taxa
2. Novel Phage Discovery
| Approach | Methodology |
|---|---|
| Similarity Screening | BLAST-based novelty assessment |
| Diversity Profiling | Environmental/host-specific virome characterization |
| Functional Gene Mining |
- Known functions: Lyases, polymerases, terminases, host-range determinants
- Unknown functions: PHROGs clustering + conserved domain identification
- CRISPR Systems
- Host spacer matching (historical infections)
- Novel anti-CRISPR (ACR) gene discovery
3. Phage Therapy Development
- Candidate Screening: Pathogen-targeted phage identification
- Host Range Prediction: Receptor-binding protein analysis + infection data correlation
- Safety Assessment: Lysogeny/virulence/antibiotic resistance gene screening
- Biocatalyst Discovery
- Therapeutic enzymes (endolysins)
- Specialized DNA polymerases
4. Metagenomic Analysis Foundation
- Reference Framework
- Binning and taxonomic assignment
- Functional annotation benchmark
- Virome Exploration
- Ecosystem studies:
- Gut microbiomes
- Marine environments
- Soil communities
- Ecosystem studies:
5. Comparative Genomics Insights
- Modular Evolution
- Conservation/recombination analysis of functional units:
- DNA replication modules
- Structural protein clusters
- Packaging systems
- Host lysis machinery
- Conservation/recombination analysis of functional units:
Multifunctional Validation of Phage Database Capabilities
1. Novel Phage Discovery & Classification
- High-Sensitivity Detection: Identified novel Rhizobium RR1-like phages with <30% sequence similarity to known entries via BLAST/BLAT
- Taxonomic Expansion: Cataloged 733 phages across 51 families using core gene clustering and ICTV standards
2. Non-Model Host Applications
- Case Study: Wild Banana (M. balbisiana) & Rare Genotypes (M. sikkimensis)
- Detected unprecedented phage diversity
- Validated database capacity for atypical host-phage systems
3. Host-Virus Interaction Analysis
| Finding | Database Function Validated |
|---|---|
| Minimal phage community overlap between banana genotypes | Host-specific association screening |
| Rhizobium-endophyte phage linkages | Taxonomy ID cross-referencing (NCBI) |
4. Functional Gene Mining
- Virulence Factor Detection: Identified holin (lysis) and Shiga-toxin gene fragments via VFDB/PHROGs integration
- Therapeutic Potential: Discovered Klebsiella phages with Fusarium wilt suppression markers through antiSMASH metabolite analysis
5. Niche Adaptability Profiling
- Tissue-Specific Distribution
- Leaf niche: Higher phage abundance (ENVO:00005784 "phyllosphere")
- Root niche: Greater diversity (Shannon Index >4.2)
- Endogenous vs. Transient Virus Discrimination
- Classified 56 Badnavirus strains using "Plant endophyte" vs. "Environmental parasite" tags
6. Knowledge Gaps as Discovery Catalysts
- Annotation Deficits
- 1,038 uncharacterized protein domains reveal novel viral lineages (Aghdam SA et al., 2023)
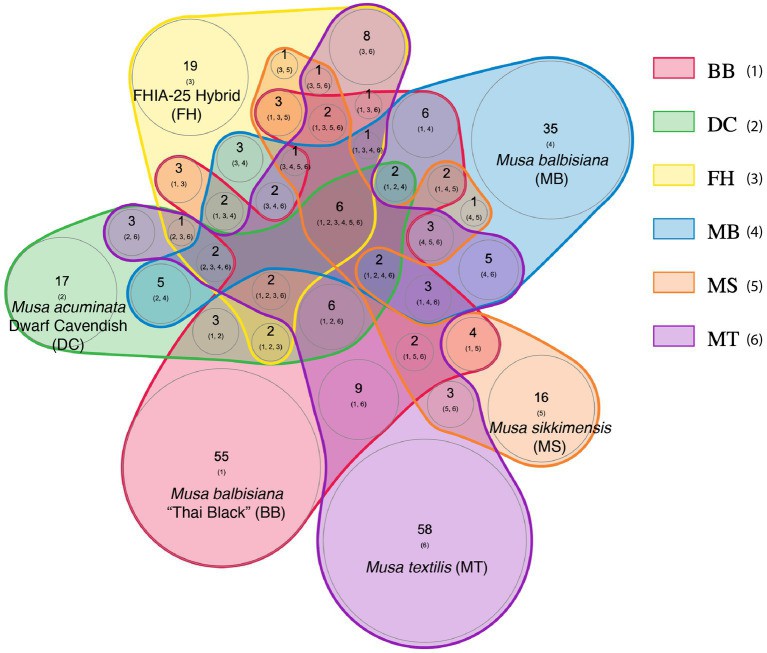 Putative phage community overlap at the lowest taxonomic-levels (species or isolates) within the endosphere microbiomes of 6 Musa genotypes (Aghdam SA et al., 2023)
Putative phage community overlap at the lowest taxonomic-levels (species or isolates) within the endosphere microbiomes of 6 Musa genotypes (Aghdam SA et al., 2023)
Explore our Service →
Challenges and Future Directions
Persisting Challenges
- Despite substantial advances, key limitations remain:
- Data Quality Gaps: Inconsistent standardization across datasets
- Host Representation Bias: Scarce genomes from non-model hosts (e.g., unculturable environmental bacteria)
- Metadata Integration Barriers:Limited interoperability of ecological/experimental context data
- Functional Knowledge Deficits: Viral "dark matter" (uncharacterized genes) representing >70% of predicted ORFs
Strategic Development Priorities
- Intelligent Annotation Systems
- AI-assisted platforms combining:
- Automated prediction pipelines
- Expert curation interfaces
- AI-assisted platforms combining:
- Phage-Host Interaction Atlas: Experimentally validated host range databases
- Multi-Omics Resource Integration
Unified access to:
| Data Type | Application |
|---|---|
| Transcriptomic | Expression dynamics |
| Proteomic | Structural verification |
| Metabolomic | Therapeutic potential screening |
- Predictive AI Implementation
- Deep learning models for:
- Gene function elucidation
- Host range projection
- Deep learning models for:
Conclusion
Phage genome databases have fundamentally transformed viral research by:
- Solving Data Management Challenges
- Enabling efficient organization of exponentially growing sequence data
- Accelerating Discovery Applications
- Serving as critical infrastructure for:
- Novel antibacterial therapies
- Synthetic biology tools
- Ecological modeling
- Serving as critical infrastructure for:
As sequencing technologies advance and global datasets expand, these repositories will remain indispensable for unlocking the full biotechnological potential of phages. Their continued evolution promises unprecedented insights into viral diversity, host adaptation mechanisms, and therapeutic engineering pathways.
Related Database Access:
PhageScope: https://phagescope.deepomics.org
PhagesDB: https://phagesdb.org
MGV: https://img.jgi.doe.gov/mgv
PhageScope:https://phagescope.deepomics.org
People Also Ask
What is the database for bacteriophages?
Welcome to PhageScope! PhageScope is an online bacteriophage database that offers comprehensive annotations, including completeness assessment, phenotype annotation, taxonomic annotation, structural annotation, functional annotation, and genome comparison.
What is a pham in phages?
Mycobacteriophage genes related to each other can be grouped into phamilies (phams) and that mosaic relationships can be analyzed and represented using pham-annotated genomes maps and phamily circles that show the patterns of which phages contain members of particular phams.
What is the ICTVdB the universal virus database?
The International Committee on Taxonomy of Viruses database is a universally available taxonomic research tool for understanding relationships among all viruses.
References:
- Fujimoto K. Metagenome data-based phage therapy for intestinal bacteria-mediated diseases. Biosci Microbiota Food Health. 2023;42(1):8-12.
- Wang RH, Yang S, Liu Z, Zhang Y, Wang X, Xu Z, Wang J, Li SC. PhageScope: a well-annotated bacteriophage database with automatic analyses and visualizations. Nucleic Acids Res. 2024 Jan 5;52(D1):D756-D761.
- Aghdam SA, Lahowetz RM, Brown AMV. Divergent endophytic viromes and phage genome repertoires among banana (Musa) species. Front Microbiol. 2023 Jun 9;14:1127606.
- Gauthier CH, Cresawn SG, Hatfull GF. PhaMMseqs: a new pipeline for constructing phage gene phamilies using MMseqs2. G3 (Bethesda). 2022 Nov 4;12(11):jkac233.
- Wang RH, Yang S, Liu Z, Zhang Y, Wang X, Xu Z, Wang J, Li SC. PhageScope: a well-annotated bacteriophage database with automatic analyses and visualizations. Nucleic Acids Res. 2024 Jan 5;52(D1):D756-D761.


