
 Sample Submission Guidelines
Sample Submission Guidelines
Quality Control for Phage Display Libraries with NGS Data
The success of phage display screening hinges fundamentally on library quality. Traditional quality control—relying on low-throughput Sanger sequencing and functional spot checks—fails to comprehensively assess compositional diversity and integrity.
Phage Next-generation sequencing (NGS) transforms this paradigm by enabling molecular-level characterization through high-throughput deep sequencing. This approach facilitates comprehensive assessment across key quality dimensions.
Key Quality Control Dimensions
1. Construction and Diversity of Phage Display Libraries
Library construction fundamentally determines phage display quality. Diversity levels directly influence both the richness and reliability of subsequent screening outcomes. Traditional diversity assessment typically relies on monoclonal amplification. In contrast, NGS technology rapidly delivers comprehensive, accurate diversity data through high-throughput sequencing of entire libraries.
NGS enables precise calculation of individual sequence frequencies, facilitating robust evaluation of library diversity and coverage. An optimal library must possess sufficient diversity to guarantee identification of high-affinity candidate peptides or proteins targeting specific molecules.
2. Quantitative Analysis and Deviation Detection
Deep sequencing via NGS quantifies the abundance of every sequence within a library. Detecting deviations is critical during quality control. Common issues include:
- Cloning Bias: Certain sequences may become disproportionately abundant due to PCR amplification artifacts. NGS analysis of relative sequence abundance identifies such enriched or biased clones.
- Indels: Insertion or deletion errors during library construction can lead to missing or truncated sequences. NGS precisely pinpoints these defects, aiding refinement of construction protocols.
 Identification of alternative binders in the NGS data set (Nannini F et al., 2021)
Identification of alternative binders in the NGS data set (Nannini F et al., 2021)
3. Library Stability and Reproducibility
Consistent library stability and batch-to-batch reproducibility are essential for reliable phage display screening results. NGS facilitates comparative analysis between library batches, ensuring consistent construction quality. Sequencing comparisons reveal whether sequence composition remains stable across batches and detect unwanted mutations or contamination.
4. Post-Screening Data Analysis
Following screening rounds, NGS data enables detailed analysis of enrichment outcomes. Researchers identify affinity-enriched sequences by analyzing shifts in sequence abundance post-screening. Key analytical steps include:
- Sequence Alignment: Comparing screened sequences to the original library identifies sequences exhibiting increased frequency (enriched) or depletion (eliminated).
- Enrichment Analysis: Tracking sequence abundance changes across successive screening rounds highlights candidate peptides or proteins progressively enriched as high-affinity binders.
5. Data Quality Assessment and Error Control
NGS data quality critically impacts result reliability. Rigorous assessment during library QC involves:
- Sequencing Depth: Ensuring sufficient coverage to accurately detect all library sequences.
- Data Accuracy: Validating accuracy by comparing results against known standards. Removing low-quality sequence data is necessary to maintain analytical reliability.
Structural Integrity Verification
Paired-end sequencing (2 × 150 bp) was employed to cover the insert and its flanking regions. Through read assembly, the following critical elements were authenticated: the integrity of the signal peptide (e.g., PelB); maintenance of the correct translational reading frame (absence of frameshift errors); purity of the CDR regions (free of stop codons); and the precise sequence of functional tags (e.g., His-Tag/Myc epitope).
Diversity Quantification and Bias Diagnosis
Core quality metrics encompass several criteria. The library's effective size, represented by the number of functional clones, must surpass 10% of its theoretical diversity. A Shannon Diversity Index exceeding 8 (on a log₁₀ scale) is required to confirm adequate distribution evenness. Sequence fidelity demands that position-specific mutation rates and codon usage exhibit less than a 15% deviation from the designed specifications. Additionally, in-depth coverage analysis is necessary for key functional regions.
Assessments Specific to Library Type
1. Antibody/Nanobody Libraries:
Evaluation includes verifying the coverage of CDR mutations (e.g., ensuring the CDR-H3 length distribution aligns with the design), analyzing the frequency of germline gene usage to detect unintended amplification bias, and screening for potential stability risks such as aggregation-prone hydrophobic patches, unpaired cysteine residues, or undesirable glycosylation motifs.
2. Peptide Libraries:
Assessment involves detecting aberrant enrichment of secondary structures (e.g., α-helix/β-sheet) using Position-Specific Scoring Matrix (PSSM) analysis. The integrity of crucial functional motifs, such as protease cleavage sites and the capacity for disulfide bond formation, is also evaluated.
Functional Prescreening and Structural Prediction
Integrated computational approaches are utilized for in silico analysis. For instance, conformational simulations are performed on NGS-derived CDR3 sequence sets (e.g., 10,000 variants) using tools like NanoNet or RoseTTAFold. These simulations predict the topology of binding pockets (classifying them as convex, concave, or flat). Furthermore, clones are screened for electrostatic complementarity to the target's epitope charge distribution.
Standardized Quality Control Workflow
A phased implementation of QC is recommended:
- Post-Construction: A minimum of 1 million reads is required to evaluate the insertion rate, reading frame integrity, and presence of stop codons.
- Post-Amplification: At least 5 million reads are needed to assess the diversity index and identify any clonal preferences.
- Pre-Panning: A functional conformation assessment should be conducted on a minimum of 10,000 representative sequences.
More information on next-generation sequencing-enhanced phage libraries can be found here "Phage Display and NGS: How Next-Generation Sequencing Enhances Phage Libraries" .
To see how the Illumina platform can deep sequence phage libraries, see "Deep Sequencing of Phage Libraries Using Illumina Platforms".
Comprehensive Case 1: NGS-Guided Quality Control in Peptide Selection
Establishment of Baseline Library Characterization
Sequencing of the initial naïve library established a reference profile for amino acid frequencies and their positional preferences (e.g., serine at 10.8%, L-Cysteine maintained below 1%). This analysis also identified an N-terminal arginine/lysine frequency gradient, with the lowest occurrence at position 1 increasing to the highest at position 12.
Verification of Library Integrity
Analysis of 521,981 sequences confirmed a high level of fidelity, with over 95% congruence between the synthesized sequences and the designed codons. The assessment also detected structural anomalies, including aberrant stop codons (leading to premature termination) and frameshift mutations, through alignment of flanking sequences.
Dynamic Monitoring of the Screening Process
NGS was employed to track the molecular evolution of 12 components across three biopanning rounds. Targeted enrichment was evident: the frequency of the QxQ motif in eluted fractions surged from 0.21% to 26.85%. This enrichment was even more pronounced in the strong elution (stripping) fraction, where the C-terminal QxQ motif reached 92.6%. Concurrently, the persistence of the HxH motif in wash fractions indicated a nonspecific binding preference.
Quantification of Amplification Bias
A comparative analysis of pre- and post-amplification libraries revealed a shift in amino acid composition. There was an increased frequency of polar residues (Thr/Ser) and a decrease in hydrophobic residues (Val/Ile). A notable reduction in cysteine frequency—from 0.99% to 0.36%—suggested phage inactivation potentially mediated by disulfide bond formation.
Explore our Service →
Key Quality Control Outcomes
1. Functional Motif Validation:
pLogo and MEME analysis localized the C-terminal QxQ (positions 10-12) as the critical arsenic-binding motif. Its frequency escalated from 0.14% to 69.23% throughout the screening rounds. Independent database interrogation (48HD.Cloud) confirmed that QxQ is a non-dominant motif in natural libraries, ranking around ~4500th.
2. Elimination of False Positives:
Application of a cross-set analysis algorithm (ES = (∩ elution ∪ ∩ stripping) ∩ (∩ washing ∩ ∩ input)) effectively filtered out rapidly proliferating clones (e.g., those containing the HxH motif). This process identified 13 high-confidence target peptides, 9 of which contained a conserved xxMPxTxxGQVQ motif.
3. Traceability of Defects:
NGS detected the post-amplification depletion of hydrophobic aromatic residues (Phe/Tyr). The root cause was traced to aggregation loss occurring during the PEG/NaCl precipitation step.
Value of the Technological Breakthrough
- Efficiency: NGS captured the library's diversity by detecting 46 unique sequences (43 of which were single-occurrence), a task that required traditional monoclonal validation of 68 clones to find only 3 repeats.
- Novel Insight: The analysis revealed N-terminal positional effects, including a complete absence of proline at position 1 (attributed to steric hindrance) and a reduced frequency of glutamate at positions 1-4 (where negative charge impedes target binding).
- Design Guidance: These findings directly informed the design of subsequent libraries, recommending the incorporation of a C-terminal L-Glutamine doublet (QxQ) and the avoidance of N-terminal negatively charged residues (Braun R et al., 2020).
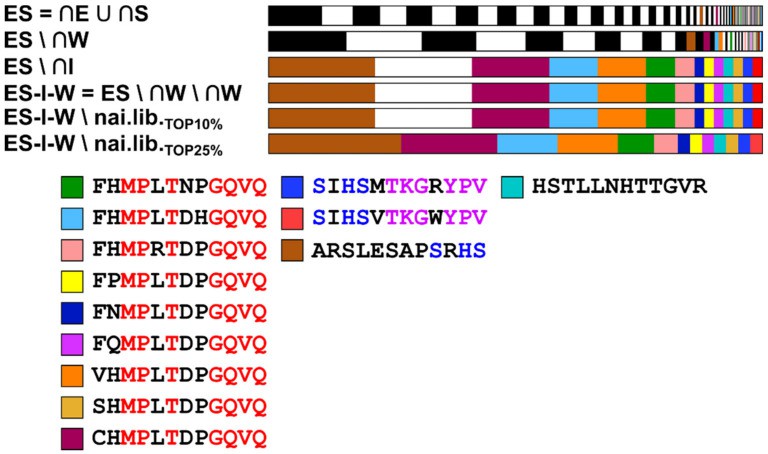 Visualization of the relative frequency of the unique sequences in the core fraction ES–I–W\naï.lib (Braun R et al., 2020)
Visualization of the relative frequency of the unique sequences in the core fraction ES–I–W\naï.lib (Braun R et al., 2020)
Comprehensive Case 2: NGS Detection of Phage Display Library Corruption
1. Precise Corruption Diagnosis
- Dominant Null Clone Exposure: Ultra-deep NGS (25M reads) revealed 95% of third-round eluate phages displayed identical non-functional peptide WSLGYTG. Conventional Sanger sequencing (30 clones) completely missed this signal.
- Growth Advantage Mechanism: Whole-genome sequencing identified lacZ gene deletion, conferring 142× amplification efficiency over normal clones, driving library homogenization collapse.
2. Diversity Decay Dynamics
- Monoclonal Collapse: Healthy initial diversity (41% monoclonals) deteriorated to 0.04% in eluates, indicating severe amplification bottlenecks.
- Functional Sequence Loss: Unique sequences decreased 770-fold (61.6% → 0.08%), confirming competitive exclusion of low-frequency high-affinity clones by growth-dominant variants.
3. False-Positive Mechanism Analysis
- Amplification-Driven Artifacts: WSLGYTG clones proliferated identically in target-free controls, confirming enrichment stemmed from amplification dominance (Pr-TUP), not binding capability.
- Sanger Sequencing Limitations: While Sanger detected 11 false-positives, NGS revealed their true rankings >1000th, exposing 90% sampling bias in traditional methods.
4. Screening Failure Attribution
- Recovery Rate Illusion: 200-fold output increase (1.6×10⁻⁶→3.4×10⁻⁴) across screening rounds represented corrupt clone amplification, not genuine target enrichment.
- Functional Invalidation: SPR and flow cytometry confirmed candidate peptides (e.g., p16/p18) lacked binding activity, corroborating NGS corruption diagnosis.
Technical Value & Quality Control Implications
- NGS enables:
- Corruption Detection: Ultra-deep scanning (0.08% sensitivity) → growth-dominant clone tracing → diversity decay quantification
- Critical Revelations:
Primary screening failures originate from library corruption, not target issues or design flaws
Recovery rate is unreliable; monoclonal proportion (>20%) should assess screening health
Sanger sequencing introduces severe distortion; NGS is essential for precise mining (Sell DK et al., 2024)
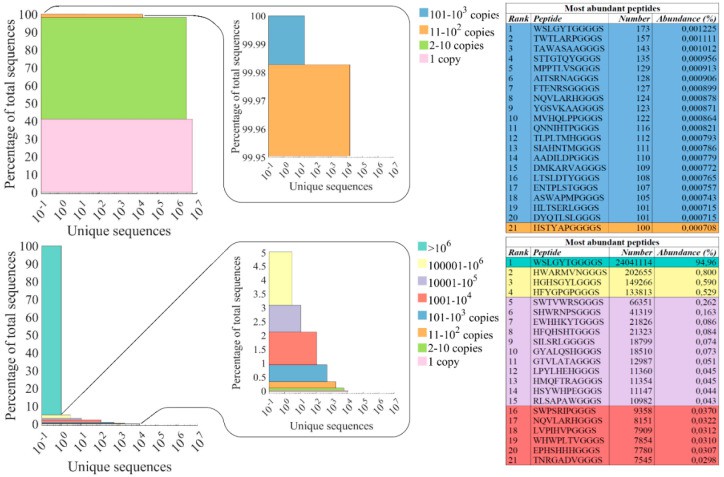 The abundance of clones in the naïve Ph.D.TM-7 library (top) and the 3rd round eluate (bottom) are presented as stacked bar plots (Sell DK et al., 2024)
The abundance of clones in the naïve Ph.D.TM-7 library (top) and the 3rd round eluate (bottom) are presented as stacked bar plots (Sell DK et al., 2024)
Comprehensive Case 3: Accelerating Target-Specific Peptide Discovery via NGS
1. Library Construction Quality Control
- Sequence Integrity Verification:
- Full-length antibody gene coverage (1.4 kb) using long-read NGS (e.g., PacBio)
- Detection of frameshifts/stop codons
- Functional clone rate increased to >95% (vs. 70% with Sanger sequencing)
- Diversity Quantification:
- Shannon Index >9.0 (indicating >10¹⁰ library capacity)
- Identified codon usage bias (e.g., SER design: 33% → CDR3 measurement: 62%)
- Guided synthesis protocol optimization
2. Screening Process Monitoring
- Corruption Early Warning:
- Real-time alerts when high-frequency invalid clones (e.g., WSLGYTG) exceed 95%
- Prevents resource waste (e.g., CD4-targeted screening: 90% cost savings)
- Functional Motif Tracking:
- Quantified target motif enrichment (e.g., arsenic-binding QxQ: 0.14% → 69.23% over 3 rounds)
- Confirmed screening efficacy
3. Deep Functional Clone Mining
- Low-Abundance High-Affinity Recovery:
- NGS detected 54% of high-affinity antibodies (EC₅₀ <0.1 nM) missed conventionally
- Exclusive identification of 229E virus neutralizing antibodies
- Germline Bias Correction:
- Revealed unexpected VH1-69/VK1-39 enrichment (44.2%)
- Guided rational CDR mutagenesis
4. Affinity Maturation Guidance
- Non-Greedy Optimization:
- ML models trained on NGS datasets enabled polyclonal parallel mutagenesis
- Discovered cryptic high-potential variants (100× affinity increase)
- Structure-Activity Prediction:
- NanoNet simulated 10,000 CDR3 conformations
- Identified 14-aa bend topology (>95% prevalence)
- Tripled success rate for concave epitope targeting
Technology Breakthrough Value
NGS transforms quality control from spot-checking to full-cycle monitoring through:
| Innovation | Application |
|---|---|
| Corruption Index (High-frequency % / Diversity decay slope) | Early screening health warning |
| Functional motif enrichment heatmaps | Dynamic wash condition optimization |
| Conformational fitness metrics (e.g., CDR3 bend ratio) | Binding activity prediction |
Establishes novel molecular QC paradigms for undruggable target development (Bakhshinejad B et al., 2025)
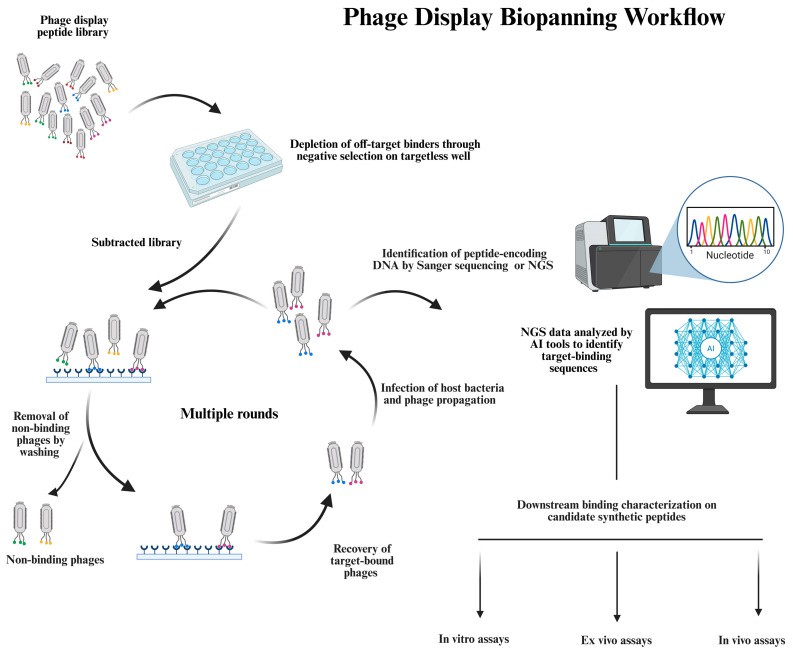 A schematic overview of the phage display biopanning workflow and downstream validation (Bakhshinejad B et al., 2025)
A schematic overview of the phage display biopanning workflow and downstream validation (Bakhshinejad B et al., 2025)
Conclusion
NGS Quality Control transforms phage library development from a "Black box operation" to a data-driven fine control process. Through the three-layer verification system of sequence integrity → diversity → functionality, it not only avoids the risk of screening failure, but also provides a molecular blueprint for the rational design of the next generation of highly active libraries. With the integration of long-read technology and AI prediction model, library quality control is moving towards a full closed-loop intelligent era of "Design-construction-verification".
For more information on how to construct and use phage Sequence database, please refer to "Building and Using Phage Genome Sequence Databases".
People Also Ask
What are the limitations of phage display?
Phage display may not recover all antigen-specific mAbs present in a given antibody library. The heavy- and light-chain pairing may not reflect that of the in vivo immunoglobulin.
What is library panning?
In its simplest form, panning is carried out by incubating a library of phage-displayed peptides with a plate (or bead) coated with the target, washing away the unbound phage, and eluting the specifically bound phage.
What is phage biopanning?
Phage display biopanning is an important, widely used tool to identify and isolate specific positive clones from large antibody fragment libraries and develop initial candidates through various further engineering strategies, such as in affinity maturation.
What is phage control?
Phage therapy is a kind of biological control where bacteriophages are employed to control microbial populations instead of antibacterial agents. Bacteriophages interact with specific receptors on the host membrane and then inject their genetic material into bacteria.
What are the limitations of phage typing?
Phage typing requires the use of a comprehensive number of phages, so it is typically only used in reference laboratories. It also relies on the interpretation of the individual lysis pattern and comparison to a standard which has led to conflicting results from different laboratories in the past.
References:
- Nannini F, Senicar L, Parekh F, Kong KJ, Kinna A, Bughda R, Sillibourne J, Hu X, Ma B, Bai Y, Ferrari M, Pule MA, Onuoha SC. Combining phage display with SMRTbell next-generation sequencing for the rapid discovery of functional scFv fragments. MAbs. 2021 Jan-Dec;13(1):1864084.
- Braun R, Schönberger N, Vinke S, Lederer F, Kalinowski J, Pollmann K. Application of Next Generation Sequencing (NGS) in Phage Displayed Peptide Selection to Support the Identification of Arsenic-Binding Motifs. Viruses. 2020 Nov 27;12(12):1360.
- Sell DK, Bakhshinejad B, Sinkjaer AW, Dawoodi IM, Wiinholt MN, Sloth AB, Stavnsbjerg C, Kjaer A. Using NGS to Uncover the Corruption of a Peptide Phage Display Selection. Curr Issues Mol Biol. 2024 Sep 21;46(9):10590-10605.
- Bakhshinejad B, Ghiasvand S. A Beautiful Bind: Phage Display and the Search for Cell-Selective Peptides. Viruses. 2025 Jul 12;17(7):975.
- Altendorf T, Mohrlüder J, Willbold D. TSAT: Efficient evaluation software for NGS data of phage/mirror-image phage display selections. Biophys Rep (N Y). 2024 Sep 11;4(3):100166.


