
 Sample Submission Guidelines
Sample Submission Guidelines
GWAS in Agriculture and Plant Breeding: Applications in Gene Mapping and Crop Improvement
Crop improvement is the core means to ensure food security, cope with climate change, and meet the needs of global population growth. According to the forecast of FAO, the global grain output needs to be increased by 50% by 2050 to meet the growing population demand, and the frequent extreme weather caused by climate change makes it increasingly difficult for crops cultivated by traditional breeding methods to adapt to the complex environment.
With the rapid development of molecular biology and genomics technology, genome-wide association study (GWAS) has become a key tool to analyze the genetic basis of complex traits of crops and accelerate the breeding process by virtue of its advantages, such as high capacity, no need to construct a specific population, and simultaneous analysis of multiple traits. By scanning single-nucleotide polymorphism (SNP) sites in the whole genome, GWAS can mine the genetic variation associated with the target trait. Compared with the traditional quantitative trait locus (QTL) mapping method, GWAS can break through the linkage disequilibrium and achieve more precise gene mapping.
The article discusses how GWAS serves as a key tool in agriculture and plant breeding, covering its applications in crop improvement, integration with QTL mapping, challenges in plant GWAS, and its future in sustainable agriculture.
GWAS for Crop Improvement
By analyzing the association between genomic markers and phenotypic traits of a large number of individuals in natural populations, GWAS can efficiently mine genetic loci that control complex traits and provide accurate molecular targets for crop improvement. Its application in crop improvement is mainly reflected in the following aspects:
Genetic Analysis of Complex Agronomic Traits
For complex traits such as yield, quality, and stress resistance (such as drought resistance and disease resistance) controlled by multiple genes, GWAS can locate key mutation sites in the whole genome. In the study of yield traits, through the GWAS analysis of the 1000-grain weight of wheat, researchers scanned thousands of wheat germplasm resources with high-density SNP chips, and combined with many years of field phenotypic data, successfully located the TaGW2 gene locus on chromosome 3B, and the variation of this locus can explain the phenotypic variation of 12%-18%, providing an important molecular target for high-yield breeding of wheat.
In terms of quality traits, cotton fiber length is an important index that affects the cotton spinning industry. By integrating transcriptome and metabolomics data, GWAS research has located a number of key QTLs related to fiber elongation in island cotton, among which the GhMML4 gene is highly expressed in the fiber elongation period, regulating the cellulose synthesis pathway, and related markers have been applied to molecular marker-assisted breeding of high-quality long-staple cotton.
 Bridging the gap: Lipidomics-assisted GWAS (lGWAS) (Pranneshraj et al., 2022)
Bridging the gap: Lipidomics-assisted GWAS (lGWAS) (Pranneshraj et al., 2022)
Target Development of Molecular Marker-assisted Breeding
The significant association markers found by GWAS can be directly used as screening tools for molecular breeding to realize early selection of excellent characters. This technical breakthrough has fundamentally changed the traditional model of crop breeding relying on phenotypic observation. Genetic loci closely linked with target traits are directly located by Qualcomm quantitative genotyping technology, which enables breeders to accurately screen individuals carrying excellent genes in the early growth stage of crops and even in the seed stage.
Compared with traditional phenotypic selection, assisted selection based on GWAS markers has significant advantages: firstly, it can effectively overcome the interference of environmental factors on phenotypic identification and realize accurate selection at the gene level; Secondly, it can break through the time and space limitation of trait expression, such as early prediction of yield traits that can only appear in the late growth stage; Thirdly, the breeding cycle is greatly shortened. In traditional breeding, it may take 1-2 years to complete the phenotypic screening of a generation, while molecular marker-assisted selection can compress the screening time of a single generation to several weeks, significantly improving the selection efficiency.
 Results from simulations under colocalization/non-colocalization scenarios (A, B), and results from real data application (C) (Giambartolomei et al., 2018)
Results from simulations under colocalization/non-colocalization scenarios (A, B), and results from real data application (C) (Giambartolomei et al., 2018)
Services you may interested in
Learn More
The Bridge Between Gene Cloning and Functional Verification
The correlation interval of GWAS mapping defines the precise boundary for the cloning of candidate genes and establishes a solid basic framework for the follow-up gene function research. In practical research, researchers usually use a variety of cutting-edge technical means to dig deep into and verify the functions of key genes from the correlation interval.
First of all, through gene expression analysis, researchers can obtain gene expression patterns at different growth stages and under different environmental conditions, and initially screen candidate genes with potential functions; Then, with the help of advanced gene editing techniques such as CRISPR/Cas9, the candidate genes were targeted knocked out, overexpressed or modified, and their effects on phenotype were observed, to accurately determine the biological functions of the genes.
Integrating GWAS with QTL Mapping
Quantitative trait loci (QTL) mapping (based on linkage analysis) and GWAS (based on linkage disequilibrium) have their advantages in genetic analysis. Their integration can complement each other's advantages and improve the accuracy and efficiency of complex trait analysis.
Complementarity Principle and Integration Logic
QTL mapping technology is mainly applied to isolated populations with a simple genetic background, such as the F2 population and backcross population. This method can effectively identify genetic loci with significant effects, but its positioning resolution is relatively limited. In contrast, GWAS relying on natural populations to carry out research, can detect minor genetic loci and has high positioning accuracy.
However, it should be noted that the GWAS method is easily disturbed by the group structure in the implementation process. Given the advantages and disadvantages of the two methods, the comprehensive coverage of major and minor loci can be achieved by integrating QTL mapping and GWAS technology, and a systematic research framework of "QTL preliminary mapping-GWAS fine mapping" can be constructed.
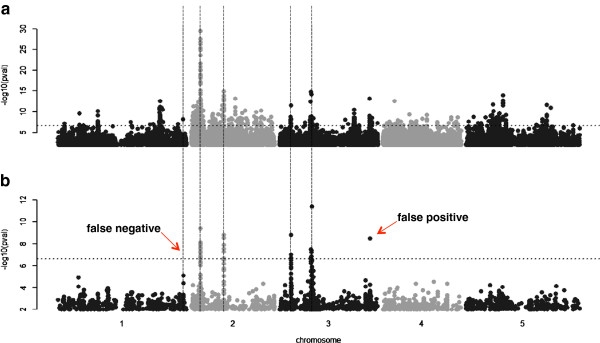 Taking genetic background into account improves the performance of GWAS (Korte et al., 2013)
Taking genetic background into account improves the performance of GWAS (Korte et al., 2013)
Technology Integration Method
Firstly, QTL mapping was used to locate the major loci in the isolated population, and then GWAS was used to scan the region carefully in the natural population to narrow the candidate interval. This strategy gives full play to the advantages of the two technologies, namely, the ability of QTL mapping to detect large-effect sites in populations with relatively simple genetic backgrounds, and the characteristics of GWAS to achieve high-resolution mapping in natural populations with rich genetic diversity.
Using statistical methods (such as mixed linear model) to directly integrate the genotype and phenotype information of the two types of data, and estimate the effects of QTL and GWAS loci at the same time. This method breaks through the limitation of the traditional single analysis method and can capture the genetic basis of complex traits more comprehensively.
The integration strategy significantly improves the accuracy of genetic locus location through multi-dimensional data fusion and algorithm optimization. The mixed linear model (MLM) and Bayesian analysis method based on machine learning can effectively control the interference caused by population structure and kinship, and improve the positioning accuracy from the chromosome segment level of traditional methods to the kb level of candidate genes.
 The current status of GWAS equipped with high-throughput phenotyping in plants (Xiao et al., 2021)
The current status of GWAS equipped with high-throughput phenotyping in plants (Xiao et al., 2021)
Challenges in Plant GWAS
Although GWAS has become an important tool for crop genetic research and breeding improvement, its practical application is still subject to multiple constraints of plant biological characteristics and research conditions, and it faces many technical and theoretical challenges that need to be solved urgently.
- The natural population of plants often has obvious subgroup differentiation (such as different geographical sources and breeding backgrounds), which can easily lead to false associations. There are great genetic differences between indica and japonica rice subspecies, and if they are directly mixed, the subspecific markers may be misjudged as loci associated with traits. In addition, the genetic basis of the same trait in different subgroups may be different (genetic heterogeneity), which increases the difficulty of analyzing the results.
- The plasticity of plant phenotype is significantly controlled by environmental covariates, including abiotic factors such as light radiation intensity, soil water content, and nutrient supply gradient. At the same time, crop yield components, quality-related indicators, and other important agronomic traits are characterized by dynamic changes and multi-dimensional composite properties, which makes the measurement errors generated in the process of phenotypic data collection easily lead to the signal attenuation of genetic effects. Taking the 1000-grain weight as an example, the accuracy of phenotypic data significantly depends on factors such as maturity judgment at harvest time, uniformity of grain development, etc. If phenotypic data collection lacks standardized operation procedures and a quality control system, it will directly lead to a significant decline in the statistical test efficiency of GWAS.
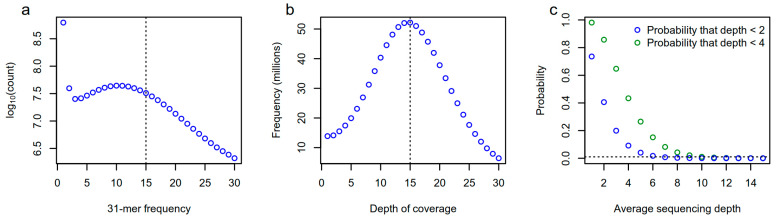 Implications of sample sequencing depth for k-mer-based GWAS (Karikari et al., 2023)
Implications of sample sequencing depth for k-mer-based GWAS (Karikari et al., 2023)
- In the field of genetic research of polyploid crops (such as potato and sugarcane), the complexity of genome structure significantly increases the research difficulty. Due to the high repetition and sequence homology of polyploid genomes, the assembly of reference genomes generally has some problems, such as poor continuity and insufficient integrity. A large number of unclosed sequence gaps and wrong annotation sites seriously interfere with the accurate positioning of molecular markers in GWAS.
At the same time, the genotyping technology based on a low-density single-nucleotide polymorphism (SNP) chip is difficult to effectively capture rare alleles and structural variation sites due to limited probe coverage. Although genome-wide resequencing technology can realize comprehensive genetic variation detection, its high sequencing cost and data processing pressure still make the deep genetic analysis of large-scale populations face double constraints in technology and economy.
Future of GWAS in Sustainable Agriculture
With technological innovation and method optimization, GWAS will play a more critical role in sustainable agricultural development, and promote the transformation of crop breeding to an efficient, accurate, and environmentally friendly direction.
- A. Combination with Environmental Adaptability Breeding
- a) GWAS is used to analyze the genetic response of crops to extreme climate stress (including high temperature, drought, salinity, and other adverse conditions), which can effectively excavate key regulatory sites and provide a theoretical basis and genetic resources for molecular design and breeding of stress-tolerant crop varieties. Based on the linkage disequilibrium principle of natural populations, this method accurately locates the QTL controlling the target traits through the correlation analysis between Qualcomm genotype and phenotypic data.
- B. Genetic Analysis of the Efficient Utilization of Resources
- a) GWAS research on the utilization efficiency of nutrients such as nitrogen, phosphorus, and potassium has become an important technical path to break through the bottleneck of agricultural resources. In the global agricultural production, the problem of environmental pollution and resource waste caused by excessive application of chemical fertilizers is becoming more and more serious, and GWAS technology can accurately locate gene loci related to key processes such as absorption, transportation, assimilation and metabolism of nitrogen, phosphorus and potassium by scanning the whole genome of large-scale natural populations.
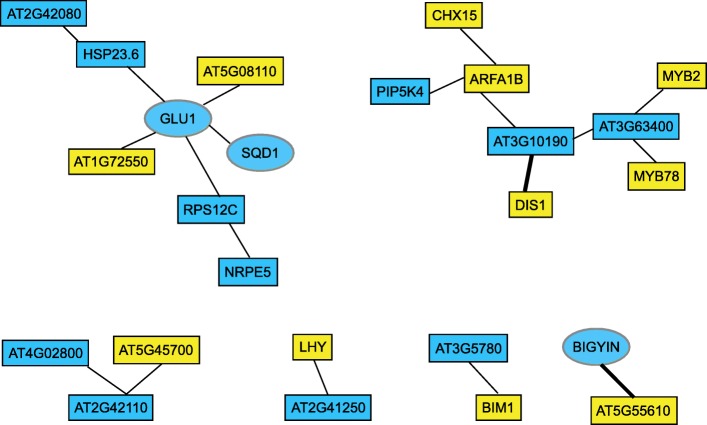 Identification of potential gene interactions from GWAS and GWES (Assefa et al., 2020)
Identification of potential gene interactions from GWAS and GWES (Assefa et al., 2020)
- C. Collaborative Innovation with Genome Editing Technology
- a) The key sites identified by GWAS can provide accurate targets for gene editing technologies such as CRISPR/Cas9, and realize directional improvement of crop characters. GWAS can efficiently locate the gene loci closely linked with the target traits by correlation analysis between phenotypic data of a large-scale population and SNP markers of the whole genome, and anchor the precise target for gene editing technology.
- b) Taking the improvement of soybean oleic acid content as an example, the researchers used GWAS to analyze the natural population of soybean containing more than 3,000 germplasm resources, and successfully located the key gene GmFAD2 controlling oleic acid synthesis. The ω-6 fatty acid desaturase encoded by this gene can catalyze the conversion of oleic acid into linoleic acid, which is the core element to regulate the degree of oil unsaturation.
Services you may interested in
- D. Cross-crop GWAS and Pan-genome Integration
- a) With the deepening of pan-genomics research, GWAS technology provides a new research paradigm for analyzing the molecular regulatory network of important agronomic traits of crops. By integrating multi-species genome variation data and systematically mining conservative genetic regulatory elements among different crops, the cross-species utilization efficiency of gene resources in crop breeding can be significantly improved.
- b) Taking Gramineae crops as the research object, through pan-genome association analysis, it was found that OsSPL14, a key gene regulating the number of grains per spike in rice, had a highly conserved biological function in maize, wheat, and other species. The research results not only provide a theoretical basis for the analysis of the conservative regulation mechanism of important agronomic traits of crops, but also lay a solid genetic foundation for the development of multi-crop cooperative improvement breeding strategies.
 Distribution of measured traits in maize under control and salt stress condition (Luo et al., 2021)
Distribution of measured traits in maize under control and salt stress condition (Luo et al., 2021)
Conclusion
GWAS has become a powerful tool for crop improvement. Through the integration with QTL mapping and the innovation of technical methods, its role in analyzing the genetic basis of complex traits and accelerating the breeding process has become increasingly prominent. Although plant GWAS still faces challenges such as population structure and phenotypic identification, with the integration of multi-omics, artificial intelligence, and other technologies, its application prospects in sustainable agriculture are broad.
In the future, GWAS will be deeply integrated with genome editing, pan-genomics, and other technologies to promote crop breeding into the era of "precise design" and provide core support for ensuring global food security and sustainable agricultural development.
References:
- Pranneshraj V, Sangha MK, Djalovic I, Miladinovic J, Djanaguiraman M. "Lipidomics-Assisted GWAS (lGWAS) Approach for Improving High-Temperature Stress Tolerance of Crops." Int J Mol Sci. 2022 23(16): 9389
- Giambartolomei C, Zhenli Liu J, Zhang W, et al. "A Bayesian framework for multiple trait colocalization from summary association statistics." Bioinformatics. 2018 34(15): 2538-2545
- Korte A, Farlow A. "The advantages and limitations of trait analysis with GWAS: a review." Plant Methods. 2013 9:29
- Xiao Q, Bai X, Zhang C, He Y. "Advanced high-throughput plant phenotyping techniques for genome-wide association studies: A review." J Adv Res. 2021 35: 215-230
- Karikari B, Lemay MA, Belzile F. "k-mer-Based Genome-Wide Association Studies in Plants: Advances, Challenges, and Perspectives." Genes (Basel). 2023 14(7): 1439
- Assefa T, Zhang J, Chowda-Reddy RV, et al. "Deconstructing the genetic architecture of iron deficiency chlorosis in soybean using genome-wide approaches." BMC Plant Biol. 2020 20(1): 42
- Luo M, Zhang Y, Li J, et al. "Molecular dissection of maize seedling salt tolerance using a genome-wide association analysis method." Plant Biotechnol J. 2021 19(10): 1937-1951


