
 Sample Submission Guidelines
Sample Submission Guidelines
Nanopore Sequencing for Structural Variation Detection, HLA Typing, and STR Analysis
In the field of human genetics, research based on whole-genome sequencing serves as the foundation for capturing essential genomic variation information, including Single Nucleotide Polymorphisms (SNPs), Insertions and Deletions (InDels), Structural Variants (SVs), Copy Number Variations (CNVs), among others. These genomic variations are considered fundamental tasks and core objectives in scientific investigation. Long read sequencing technologies, exemplified by Pacbio and Nanopore, with their distinctive feature of generating long reads, have demonstrated extensive utility in the study of human and non-human genomes.
What is Nanopore Resequencing
Nanopore resequencing is a sequencing technique that involves conducting whole-genome sequencing on human samples. Utilizing long reads ranging from 10kb to 20kb, these sequences are aligned and analyzed against the human reference genome. This process enables precise detection of genetic variations, such as Structural Variations (SVs) and Copy Number Variations (CNVs), within the DNA sequences of samples compared to the reference genome or among different samples. This technology addresses the limitations of next-generation resequencing in the detection of large-segment sequence variations.
Why Perform Nanopore resequencing
Through the exploration of SVs and CNVs, this technique finds applications in differential analyses among individuals or populations. It plays a crucial role in studies related to Human Leukocyte Antigen (HLA), Short Tandem Repeats (STRs), and other aspects in the fields of diseases and cancers. Third-generation resequencing, with its advantages of long reads (capable of spanning large structural variations, repetitive regions, high GC content, highly homologous regions, and highly polymorphic regions) and the absence of PCR amplification (avoiding errors introduced by PCR amplification), has emerged as a novel strategy for exploring genetic variation information in the human genome.
Service you may intersted in
- Nanopore metagenomics sequencing
- Nanopore de novo whole genome requencing
Resource
Variant Detection:
Genomic Structural Variations (SVs) typically refer to large-segment structural variations exceeding 50bp, encompassing various types such as deletions (DEL), insertions (INS), duplications (DUP), inversions (INV), and copy number variations (CNV). These variations, occurring at both individual and population levels, propel human genome diversity and evolution. In comparison to Single Nucleotide Polymorphisms (SNPs), SVs constitute a higher proportion of total variations and have a more widespread impact on the genome. Once changes occur, they often exert profound effects on living organisms. Mounting evidence indicates the association of SVs with numerous human diseases, including neurodevelopmental disorders, cardiovascular diseases, and cancers. Therefore, a systematic analysis of SVs in the human genome holds significant implications for both biological and clinical research.
However, the detection of SVs remains a notably challenging technological endeavor. Firstly, SVs themselves encompass a diverse array of types, typically classified into five categories: insertions, deletions, duplications, translocations, and inversions. The distinct nature of these five mutations introduces notable variations in detection complexities. For instance, deletion mutations are relatively easier to detect, whereas the identification of insertion or duplication mutations poses greater challenges. next-ly, certain SV mutations exhibit substantial magnitudes, defined as exceeding 50 base pairs, with some extending to several hundred kilobases. For short segments typical in next-generation sequencing, this presents an undeniable challenge that current technological approaches struggle to overcome. Moreover, repetitive sequences like CNVs pose additional difficulties due to the limited length of sequencing data, making them equally challenging to address.
Utilizing long-read Nanopore sequencing technology obviates the need for splicing, enabling comprehensive coverage of structural variation and/or repetitive regions, all while retaining information pertaining to polyploidy. Furthermore, by scrutinizing the sequences of both parental sources, it becomes feasible to precisely discern the occurrence of structural variations (SVs). This, in turn, furnishes more accurate and refined data for investigating the role of structural variations in diseases, evolution, and genetic diversity. Hence, at present, employing nanopore long fragments stands out as the optimal strategy for structural variation detection.
Service you may intersted in
Resource
STRs Analysis:
Short Tandem Repeats (STRs) are brief DNA sequences, typically consisting of 2 to 6 base pairs (bp), repetitively present at fixed positions in the genome. STRs constitute approximately 7% of the human genome sequence and exhibit high polymorphism, with varying lengths observed among different individuals. Abnormally elongated or "expanded" alleles of STRs represent a significant category of pathogenic variations within populations. To date, expansions of STRs in over 40 genes have been established to cause hereditary diseases. The majority of these conditions manifest primarily with neurological or neuromuscular symptoms, encompassing disorders such as Huntington's disease (HD; HTT), Fragile X Syndrome (FXS; FMR1), inherited ataxias (RFC1, FXN, etc.), myotonic dystrophy (DMPK and CNBP), muscle channelopathies (CSTB, SAMD12, STARD7, etc.), as well as C9orf72-related frontotemporal dementia and amyotrophic lateral sclerosis (ALS).
Given (i) the diverse and prevalent nature of Short Tandem Repeat (STR) expansion disorders, (ii) the multitude of involved genes, (iii) the continual discovery of new genes, (iv) the diversity in size and sequence conformation of pathogenic STR expansions, and (v) the existing gaps in our foundational understanding of their biology, there is an increasing demand for enhanced molecular detection methods for STRs. Established molecular techniques (such as Southern blotting and Repeat-Primed Polymerase Chain Reaction, RP-PCR) are relatively slow, labor-intensive, imprecise, and require individual assays with specific primers/probes for each distinct STR.
This becomes problematic when multiple different STR expansions can manifest as similar phenotypes (locus heterogeneity), posing a major obstacle to testing newly discovered STR genes. Next-generation sequencing (NGS) holds practicality in analyzing STR expansions. However, the large size, low sequence complexity, and high GC content of many pathogenic STR expansions make them challenging to analyze through short-read NGS platforms (such as Illumina). Therefore, there is a growing necessity for refined methodologies in the molecular detection of STRs.
Nanopore sequencing and Pacific sequencing, both encompassing long-read sequencing technologies, are applicable for gene typing of large and intricate STR expansions. These methodologies facilitate seamless coverage of low-complexity regions within the genome, devoid of GC bias. Another advantage of these technologies lies in the concurrent analysis of DNA methylation at repetitive loci. Hence, the utilization of third-generation long-read data for STR detection presents distinct advantages.
Service you may intersted in
Resource
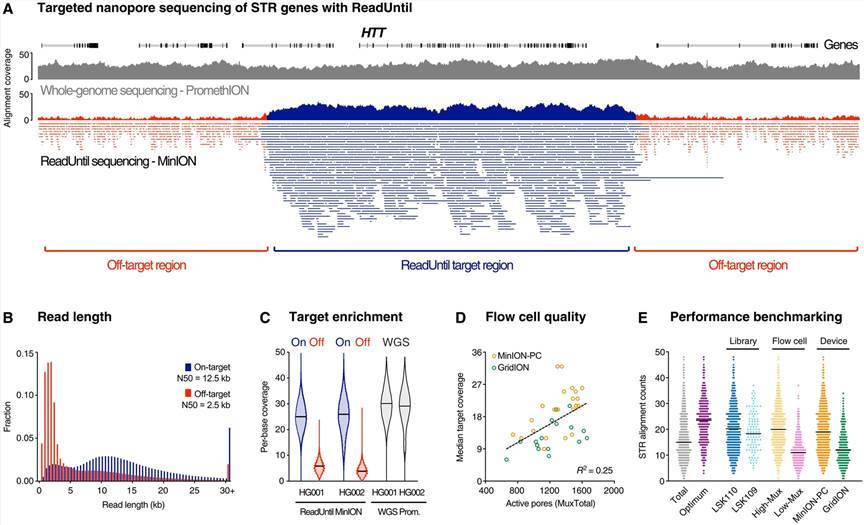 Pathogenic STR sites with ONT
Pathogenic STR sites with ONT
HLA Typing:
Human Leukocyte Antigen (HLA), located on the short arm of chromosome 6 in the human genome, comprises a series of closely linked genetic loci. Classified into three main categories—Ⅰ, Ⅱ, and Ⅲ—HLA is primarily distinguished based on the structural, functional, and cellular distribution characteristics of its gene products. The typing of Ⅰ and Ⅱ class genes holds significant relevance in clinical applications.
HLA exhibits a characteristic of high polymorphism, whereby immune cells distinguish self from non-self by recognizing HLA, making it the genetic "identity card" of humans. Closely intertwined with the body's immune system, HLA typing finds its primary applications in areas such as transplantation medicine, research on immune-related diseases, drug responses, and platelet transfusions. Given the challenging nature of accurate typing due to its polymorphic features, the precise characterization of HLA genes has consistently posed a research puzzle and a direction fervently pursued by various sequencing technologies.
Service you may intersted in
Resource
In recent years, high-throughput sequencing technologies, leveraging their advantages in throughput, comprehensive or comprehensive genomic characterization, shortened turnaround times, and reduced ambiguity in sequence determination, have supplanted Sanger sequencing as the gold standard for high-resolution typing of Human Leukocyte Antigen (HLA). This transition presents a significant advantage over conventional typing methods.
The Nanopore sequencing platform has demonstrated exceptional performance in generating long reads (thousands or millions of base pairs). Employing the "long amplicon, long read" strategy, it successfully obviates the need for DNA fragmentation during the preparation of HLA sequencing libraries. Simultaneously, it optimizes the identification of distant variations spanning thousands of base pairs. This platform achieves outstanding consistency and accuracy, widely regarded as a rapid and cost-effective solution for HLA typing.
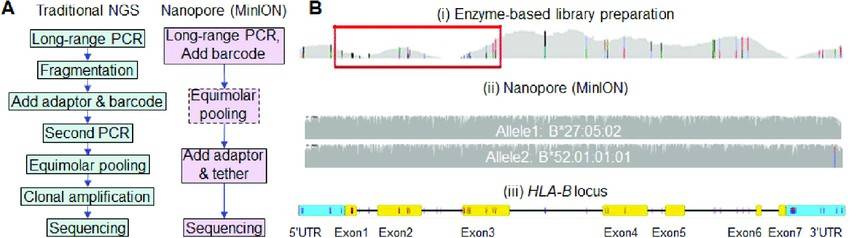 Comparison of workflows by traditional NGS and nanopore sequencing.
Comparison of workflows by traditional NGS and nanopore sequencing.
References:
- Chang Liu a., Xiao Yang b,1, Brian F Duffye, Jessica Hoisington-Lopezd, MariaLynn Crosby'Rhonda Porche-Sorbet, Katsuyuki Saito e, Rick Berry,, Victoria Swamidass', Robi D. High-resolution HLA typing by long reads from the R10.3 Oxfordnanopore flow cells. Mitra Human Immunology 2021
- Igor Stevanovski et al., Comprehensive genetic diagnosis of tandem repeat expansion disorders with programmable targeted nanopore sequencing. Sci. Adv.8,eabm5386(2022).
- Liu C. A long road/read to rapid high-resolution HLA typing: The nanopore perspective. Hum Immunol. 2021 Jul;82(7):488-495.


