
 Sample Submission Guidelines
Sample Submission Guidelines
How to Detect Structural Variations (SVs) By Sequencing
What is Structural Variation?
Structural Variations (SVs) refer to mutations within DNA segments longer than 50 base pairs. Notable SV types encompass deletion, duplication, insertion, inversion, and translocation.
Regarding their impact on gene expression, SVs can exert diverse effects:
- Variants like gene duplications, insertions, and deletions can modify gene dosage.
- Structural variations within coding regions can influence gene transcription and translation.
- SVs in non-coding regions can perturb gene regulatory elements through positional effects.
- Deletion of enhancer or repressor elements can alter gene transcription levels.
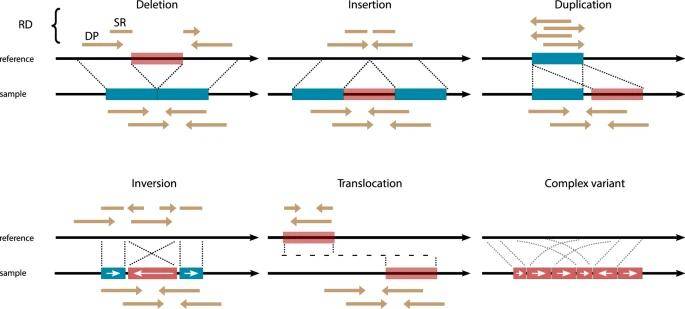 Major SV types and their characteristic read-alignment patterns. (van Belzen et al., 2021)
Major SV types and their characteristic read-alignment patterns. (van Belzen et al., 2021)
In the context of diseases, SVs may induce abnormal trait expression, leading to various hereditary conditions.
While the prevalence of structural variants in the human genome accounts for roughly 0.5% compared to single nucleotide variants (SNVs), they affect ten times more bases than SNVs in total. Structural variants are more likely to be associated with genome-wide association signals and have a higher propensity to impact gene expression compared to SNVs.
SVs stand as a major contributor to genetic disorders in humans. However, current clinical testing predominantly relies on conventional cytogenetic methods. To comprehensively detect chromosomal abnormalities, a combination of cytogenetic techniques such as fluorescence in situ hybridization (FISH), karyotyping, and copy number variation (CNV) microarrays is often necessary.
High-throughput sequencing techniques yield short nucleotide sequences, typically spanning tens to hundreds of bases, termed sequencing fragments or "reads." Most SV detection methods based on sequencing technologies identify these fragments by aligning them to a reference genome. Nevertheless, precise SV detection presents substantial challenges, given that SVs tend to reside in repetitive or duplicated DNA regions and encompass a variety of types, including insertions, deletions, inversions, translocations, duplications, further complicating their detection process.
Strategies for Detecting Structural Variants in Next-Generation Sequencing (NGS) Data
Read-Pair Method (RP)
The read-pair method, also known as paired-end sequencing (PE), involves sequencing both ends of the same DNA fragment, referred to as read1 and read2, in opposite directions. These reads are typically short, often less than a few hundred base pairs. The space between them is called the insertion fragment, with its size referred to as the insert size. Measuring the true insert length between read1 and read2 is indirect and involves aligning them to a reference genome.
PE sequencing provides valuable information about the distance and orientation of paired reads at the ends of a DNA fragment. By comparing these reads to a reference genome, we can analyze their positional and orientation information to identify structural variants (SVs) that deviate from the reference genome.
Limitations:
- The read-pair method faces challenges when dealing with repetitive genomic regions.
- It is less effective at detecting SVs in large segments due to limitations in DNA fragment length.
- Detection reliability and accuracy decrease for small deletions (typically <200bp).
- In cases where the entire insertion fragment is a sequence variation, genomic information may not be obtained.
Split Read Method
The read-depth method detects duplications and deletions by assuming a random distribution of mapping depth, such as a Poisson distribution. It identifies duplicated regions with high read depth and deleted regions with low read depth when compared to a reference genome.
Read Depth Methods
Split read methods are adept at detecting deletions and small insertions, especially with long Sanger sequence reads. These methods aim to pinpoint structural variant breakpoints and can also detect mobile insertions if the reads are sufficiently long (>400bp). However, the prevalence of short reads in NGS-based sequencing complicates alignments and limits split-read method applicability. The Pindel algorithm, for instance, leverages end-pairing reads to reduce the complexity of short sequence alignments.
Sequence De Novo Assembly (AS) Approach
In theory, de novo assembly can accurately detect all types of structural variations. However, NGS technologies primarily yield short read sequences (typically 30 bp), posing a significant challenge for whole-genome de novo assembly. Traditional assembly methods require identifying overlapping regions, increasing the complexity of assembly. Although paired-read assembly is theoretically easier, in practice, it is more complex compared to unpaired-read assembly. Various algorithms aim to combine de novo assembly with localized assembly techniques to address these challenges.
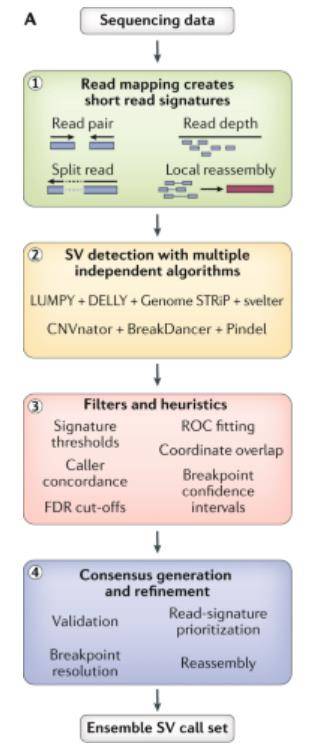 Short-read sequencing workflow to identify structural variants. (Ho et al., 2020)
Short-read sequencing workflow to identify structural variants. (Ho et al., 2020)
Long-Read Sequencing Technology Is Revolutionizing Structural Variant Detection
Long-read sequencing technology (mainly from PacBio and Oxford Nanopore) heralds a paradigm shift in structural variant detection, ushering in heightened efficiency and data fidelity. Within the realm of genomic structural variant identification, both long-read sequencing and de novo assembly stand out as formidable tools. These methods excel in delivering a comprehensive inventory of structural variants of all types. However, the intrinsic capacity of long-read sequencing equipment to generate extended DNA sequences confers a marked enhancement in detection accuracy, enabling the discernment of even the most substantial structural variants, including those elusive regions that elude detection by other technological means.
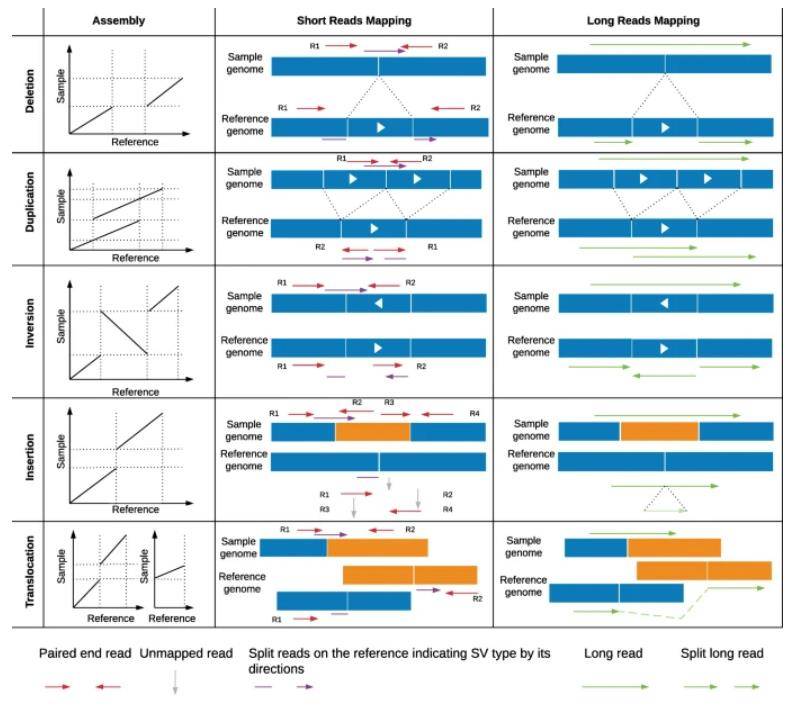 Comparison between de novo assembly, short-read and long-read mapping approaches to identify structural variants. (Mahmoud et al., 2019)
Comparison between de novo assembly, short-read and long-read mapping approaches to identify structural variants. (Mahmoud et al., 2019)
Profound Advancements in Diverse Structural Variant Detection
The long-read sequencing technology has wrought profound transformations in structural variant detection by virtue of its capacity for generating lengthy sequence reads. It not only excels in uncovering common structural variants but also adeptly captures intricate structural variations, tandem repeats, and transposable element insertions. This breadth of capability positions it leagues ahead of conventional methodologies for structural variant identification.
In contrast to conventional techniques, long-read sequencing technology excels in pinpointing intricate structural variants. These complexities often encompass multiple breakpoints and variant sequences that might confound traditional approaches. The extended read lengths intrinsic to long-read sequencing technology enable more precise identification and localization of these breakpoints, thus markedly elevating detection accuracy.
The technology also demonstrates a remarkable ability to discern tandem repeats—a form of structural variation that profoundly influences gene function and expression. Where traditional methods may falter in tandem repeat detection, long-read sequencing technology shines, effortlessly distinguishing between various repeat sequences and enriching the research dataset with comprehensive information.
When it comes to detecting transposable element insertions, long-read sequencing technology exhibits exceptional prowess. It offers a precise capture of these insertion events, affording researchers a deeper understanding of the genomic impact of such structural variants.
Beyond its diversity in structural variant detection, long-read sequencing technology also empowers researchers with more granular insights. It furnishes critical information, including the exact positions of variant breakpoints and complete variant sequences, which are indispensable for further research and the interpretation of variant functionality and repercussions.
Pinpointing Breakpoint Locations with Precision
Long-read sequencing technology excels in the accurate localization of structural variant breakpoint positions—a pivotal aspect of subsequent research and validation. Researchers can gain a more nuanced understanding of the precise locations where variants manifest, aiding in the unraveling of associations between variants and diseases or other biological processes.
Another distinctive edge of long-read sequencing technology lies in its capability to yield exhaustive variant sequence information. In stark contrast to traditional methodologies, which may provide only limited insight into variant sequences, long-read sequencing technology captures the entirety of variant sequences. This comprehensive dataset empowers researchers to conduct more exhaustive analyses of variant characteristics and their potential impacts.
References:
- van Belzen, Ianthe AEM, et al. "Structural variant detection in cancer genomes: computational challenges and perspectives for precision oncology." NPJ Precision Oncology 5.1 (2021): 15.
- Mahmoud, Medhat, et al. "Structural variant calling: the long and the short of it." Genome biology 20.1 (2019): 1-14.
- Ho, Steve S., Alexander E. Urban, and Ryan E. Mills. "Structural variation in the sequencing era." Nature Reviews Genetics 21.3 (2020): 171-189.


