
Combining Microbial Diversity and Metagenomic Sequencing: From Community Profiles to Strain-Level Functions
Inquiry >In microbiome research, scientists rarely ask only "who is there?" or only "what can they do?". Real projects usually need both taxonomic resolution and functional insight, and often across multiple environments or experimental conditions. That is where combining microbial diversity sequencing and metagenomic sequencing becomes much more powerful than using either technology alone. For researchers comparing amplicon-based microbial diversity sequencing services with shotgun metagenomic sequencing services, this guide explains how to design combined microbiome studies that move from community-level profiles to strain-level functional insights.
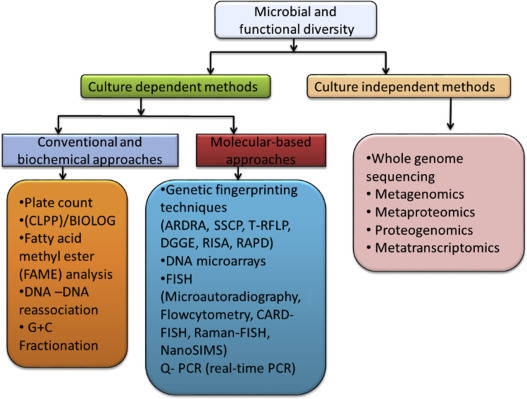
On the CD Genomics MicrobioSeq platform, more and more microbiome projects are being designed as multi-omics studies that stack 16S/18S/ITS amplicon sequencing, shotgun metagenomics, metatranscriptomics, and even metabolomics in one integrated workflow. This article focuses on how to combine microbial diversity and metagenomic sequencing in practice, how this pairing has been used in recent high-impact papers, and how to design similar research-use-only projects using CD Genomics services.
 Figure 1. Summary of how combining microbial diversity sequencing with metagenomic analysis links community composition ("Diversity") to functional genes and pathways ("Functions") in microbiome studies.
Figure 1. Summary of how combining microbial diversity sequencing with metagenomic analysis links community composition ("Diversity") to functional genes and pathways ("Functions") in microbiome studies.
1. Why Combine Microbial Diversity Sequencing and Metagenomics?
Standalone 16S/18S/ITS microbial diversity sequencing offers a fast, cost-effective way to screen many samples, identify major taxa, and track community shifts. Standalone metagenomic sequencing offers gene- and pathway-level information and can resolve genomes, but at higher cost and complexity per sample. Most ambitious microbiome questions sit in the middle: you want to understand which microbial lineages matter and what functions they carry, while still covering enough samples and conditions to be statistically robust.
Limitations of Standalone 16S/18S/ITS Amplicon Sequencing (Taxonomy Without Functions)
Amplicon-based microbial diversity analysis is ideal for:
- High-throughput profiling of many samples
- Detecting shifts in alpha and beta diversity
- Identifying taxa correlated with environmental gradients or phenotypes
But even with full-length 16S/18S/ITS, you typically infer function indirectly from taxonomy. You cannot reliably see:
- Which functional genes are present or absent
- Which metabolic pathways are being reshaped
- How strain-level variation changes ecological roles
These trade-offs are discussed in more detail in MicrobioSeq resources such as "Microbial Diversity Analysis Methods" and "Microbial Diversity: Significance and Research Methodology", which compare different amplicon approaches and emphasize how diversity metrics relate to ecosystem function.
Limitations of Standalone Shotgun Metagenomics (Cost, Depth, and Complexity)
Shotgun metagenomic sequencing directly captures all microbial DNA in a sample. You can:
- Annotate functional genes and pathways
- Recover metagenome-assembled genomes (MAGs)
- Screen for novel enzymes, biosynthetic clusters, and resistance genes
However, metagenomics alone can be limiting when:
- You need large sample series (time courses, field trials, multi-site surveys)
- Budgets constrain depth per sample
- You want simple, standardized community metrics comparable to existing diversity literature
The MicrobioSeq article "Microbial Metagenomic Sequencing: An Advancement in Ultra-High Resolution Microbiomics" explains these advantages in depth. Here, we treat metagenomics as one pillar in a combined strategy, not the whole story.
Synergy: Linking Microbial Community Structure, Function, and Ecology
In practice, integrated 16S amplicon and metagenomic sequencing lets you:
- Use amplicons to screen broadly and prioritize key samples and niches
- Use metagenomics on a subset of samples to understand mechanisms
- Map taxa ↔ functional modules ↔ environmental or host-related traits
The result is a multi-layered view of the microbiome:
- Community composition and diversity
- Absolute or relative abundance of key groups
- Functional genes, pathways, and MAG-level traits
That is the core idea behind the three joint strategies below.
 Figure 2. Three joint strategies for combining microbial diversity sequencing and metagenomic analysis: screening and zooming in with multi-omics, linking OTUs/ASVs to MAGs, and integrating absolute quantification with functional profiling.
Figure 2. Three joint strategies for combining microbial diversity sequencing and metagenomic analysis: screening and zooming in with multi-omics, linking OTUs/ASVs to MAGs, and integrating absolute quantification with functional profiling.
2. 16S/18S/ITS Amplicon vs. Shotgun Metagenomic Sequencing
Rather than re-explaining basic principles, this section focuses on what each method adds in a combined microbiome study.
 Figure 3. Conceptual comparison of 16S/18S/ITS amplicon-based microbial diversity profiling and shotgun metagenomic sequencing workflows, illustrating differences in sequencing targets, bioinformatic pipelines, and functional readouts (Liu Y.-X. et al. (2021) Protein & Cell).
Figure 3. Conceptual comparison of 16S/18S/ITS amplicon-based microbial diversity profiling and shotgun metagenomic sequencing workflows, illustrating differences in sequencing targets, bioinformatic pipelines, and functional readouts (Liu Y.-X. et al. (2021) Protein & Cell).
Microbial Diversity Sequencing for Fast Community Profiling (16S, ITS, Functional Gene Amplicons)
Microbial diversity sequencing (16S rRNA, 18S rRNA, ITS, or targeted functional genes):
- Delivers robust taxonomic profiles at the phylum–genus level (sometimes species)
- Works well for environmental microbiomes, rhizosphere, wastewater, and gut models
- Supports standardized diversity metrics (Shannon, Simpson, UniFrac, etc.)
On MicrobioSeq, this corresponds to services such as Microbial Diversity Analysis – 16S/18S/ITS Sequencing and matrix-specific solutions (soil, water, biofilms, extreme environments). These data sets are perfect for hypothesis generation: which sample groups differ, which taxa track with environmental factors, and which conditions merit deeper functional work.
You can find more technical details in our 16S/18S/ITS microbial diversity sequencing service.
Metagenomic Sequencing for Functional Gene and Pathway Discovery
In a combined design, shotgun metagenomics is usually applied to a focused subset of samples chosen based on the diversity results:
- High-contrast samples (e.g., extreme vs. moderate conditions)
- Representative stages in a time series
- Key responders identified by diversity metrics
Metagenomic analysis then reveals:
- Functional genes for CNPS cycles, metal metabolism, pollutant degradation, carbohydrate utilization, etc.
- MAGs that explain strain-level variability and metabolic potential
- Co-occurrence and functional networks that link taxa to processes
For functional profiling, our shotgun metagenomic sequencing service supports high-quality assembly, binning, and pathway annotation for complex microbiomes.
When to Add Metatranscriptomics for Active Functional Profiling
For projects where activity matters as much as potential—such as wastewater reactors at different loads, stressed rhizospheres, or host-associated models under stress—adding metatranscriptomic sequencing (RNA-based) clarifies which genes are actually expressed under each condition.
A practical pattern is:
- Amplicon → who is there
- Metagenomics → what they can do
- Metatranscriptomics → what they are doing now
This triad is particularly useful when you want to connect functional gene expression to metabolite profiles or host phenotypes in a research setting.
3. Joint Strategy 1: Microbial Diversity + Metagenomics/Metatranscriptomics
The first joint strategy is to screen with microbial diversity sequencing and zoom in with metagenomics/metatranscriptomics on the most interesting contrasts.
Screening Key Taxa with Microbial Diversity Sequencing
A typical workflow:
- Design a sampling scheme that spans gradients or treatments (e.g., drought vs. well-watered, high vs. low organic load, irradiated vs. control).
- Use 16S/18S/ITS amplicon sequencing to profile all samples.
- Cluster and ordain samples to identify community states and indicator taxa.
Two practical tips from project experience:
- Treat replication and metadata as non-negotiable. Projects with ≥3 biological replicates per group and well-annotated metadata are far easier to interpret.
- When possible, collect frozen aliquots of key samples in advance; you will need them once you decide which subset to send for metagenomic sequencing.
Validating Functional Pathways with Metagenomic / Metatranscriptomic Sequencing
Once key contrasts are chosen, metagenomic sequencing can:
- Confirm whether communities enriched in certain taxa also carry distinct functional gene sets.
- Highlight metabolic pathways that track with environment or phenotype (e.g., denitrification, sulfur oxidation, short-chain fatty acid biosynthesis, aromatic compound degradation).
- Provide MAGs that represent keystone organisms.
If you add metatranscriptomics, you gain information on which pathways are actively expressed, which is particularly helpful in dynamic systems like wastewater bioreactors or host-associated models under stress.
Mapping Microbes to CNPS Cycling, Metal Metabolism, Pollutant Degradation, and Host-Related Pathways
In many multi-omics projects, this strategy is used to link:
- Microbial diversity shifts to CNPS cycles (carbon, nitrogen, phosphorus, sulfur), especially in soils, sediments, and aquatic systems.
- Taxa enriched by pollution or metal stress to heavy metal resistance and transformation genes.
- Gut microbial community restructuring to metabolic pathways related to energy metabolism and host signaling in preclinical models.
A key design principle is to plan functional annotation ahead of time—decide early which databases (KEGG, COG, CAZy, custom gene sets) and which environmental processes you care about.
4. Joint Strategy 2: From OTUs/ASVs to MAGs
The second strategy combines community-level amplicon data with MAG-focused metagenomic analysis to move from "a group of organisms" to strain-level genomes.
Using Amplicon Data to Track Community Shifts and Candidate Taxa
Amplicon data are ideal to:
- Track temporal succession (e.g., during long-term enrichment or fermentation)
- Identify stable vs. transient members of a community
- Highlight OTUs/ASVs that correlate with performance metrics (e.g., methane yield, plant growth, pollutant removal)
These patterns guide where to invest metagenomic depth. For example:
- Time points dominated by distinct community states
- Reactors or plots with contrasting performance
- Samples where rare but important taxa become abundant
Metagenome-Assembled Genomes (MAGs) for Strain-Resolved Metabolic Potential
Once metagenomic data are assembled and binned, MAGs allow:
- Strain-resolved views of metabolic pathways, including niche differentiation among closely related taxa
- Discovery of novel lineages not represented in reference databases
- Tracing of horizontal gene transfer events and genomic islands
MicrobioSeq resources on long-read metagenomic sequencing describe how long-read platforms can improve MAG contiguity and functional interpretation in these projects.
Guiding Strain Isolation, Synthetic Microbial Consortia Design, and Colonization Validation
Pairing amplicon patterns with MAG-level data has several practical benefits:
- You can prioritize strains for isolation because you already know their genomic potential and ecological behavior.
- MAGs provide a blueprint for designing synthetic consortia (e.g., drought-tolerant rhizosphere communities, methanogenic consortia, disease-suppressive microbiomes).
- Community-level diversity sequencing becomes a readout to validate colonization and stability of synthetic consortia in subsequent experiments.
In hands-on work, it is very helpful to keep a MAG tracking table that records bin quality, taxonomy, key pathways, and which samples they came from; this becomes the backbone for downstream experimental design.
5. Joint Strategy 3: Absolute Quantification + Metagenomic Functional Profiling
The third strategy moves beyond relative abundance and uses absolute quantification of microbial diversity to better estimate ecological contributions.
Why Move Beyond Relative Abundance: eDNA and Microbial Absolute Quantification
Relative abundance can be misleading when:
- Total microbial load varies between samples
- One group blooms and mechanically decreases the relative abundance of others even if their absolute counts stay stable
eDNA absolute quantification or spike-in-based absolute quantification can correct this by providing:
- Total cell or gene copy counts per gram, milliliter, or surface area
- Absolute abundances for specific taxa or functional groups
MicrobioSeq's guide "Beyond Relative Abundance: eDNA Absolute Quantification" shows how these approaches can be integrated with microbial sequencing workflows.
Quantifying Dominant and Rare Taxa to Estimate Ecological Contribution
When absolute quantification is combined with diversity sequencing, you can:
- Measure how dominant taxa truly expand or contract in cell numbers across treatments
- Assess whether rare but functionally important groups (e.g., nitrifiers, sulfate reducers, methanogens) are increasing in real terms
- Better model ecosystem-level rates (e.g., methane production, nutrient cycling) based on measured biomass and potential
In operational terms, this usually means:
- Designing qPCR or dPCR assays for total 16S and key functional genes
- Using spike-ins or standard curves to convert relative amplicon data into absolute counts
Linking Absolute Abundance to Metagenomic Functional Modules and Metabolic Pathways
Once absolute counts are available, metagenomic functional profiles can be scaled accordingly:
- Pathway abundance can be expressed per gram or per liter, rather than as relative reads-per-million.
- You can identify "high-impact" taxa whose increase in absolute abundance drives most of the change in a pathway.
- This is particularly helpful when linking metagenomics to process-level measurements (e.g., gas production, nutrient fluxes, or host-level traits in research models).
From a practical perspective, make sure that sample volumes and extraction protocols are standardized so that per-sample absolute counts remain comparable.
6. Case Studies: Combining Microbial Diversity and Metagenomics
The following examples—aligned with published work supported by multi-omics strategies—illustrate how microbial diversity sequencing and metagenomics are combined in real research. DOIs are included so that data and methods remain traceable.
Anaerobic Wastewater Ecosystems: Methanogens with Extracellular Electron Transfer Potential
A recent Nature Water study surveyed 378 methanogen genomes and identified 84 strains with genomic potential for extracellular electron transfer (EET), including proton-pumping Fpo complexes, conductive flagella, and multiheme c-type cytochromes (DOI: https://doi.org/10.1038/s44221-025-00524-6). Amplicon data characterized community shifts across more than 500 anaerobic digestion samples, while metagenomics and comparative genomics revealed which methanogens carried EET-related genes and how they were positioned in interaction networks. This combination linked community composition and strain-level EET potential to ecosystem performance in wastewater treatment systems.
Wheat Rhizosphere: Drought-Tolerant Bacteria and Functional Taxa Enhancing Plant Resilience
In Nature Food, a global survey of the wheat rhizosphere used a combination of microbial diversity profiling and functional genomics to identify 21 highly active drought-tolerant bacteria (DTB) enriched under drought stress (DOI: https://doi.org/10.1038/s43016-025-01248-2). Amplicon profiles across the phyllosphere, rhizosphere, and root endosphere revealed shifts toward Actinobacteria and Ascomycota and depletion of Proteobacteria and Basidiomycota. Metagenomics and individual-cell genomics then linked DTB taxa to genes involved in nutrient cycling and plant resilience, supporting the design of synthetic communities that improved wheat growth under drought in experimental systems.
Gut Microbiota and Radiation Injury: Butyrate-Producing Strains Supporting Intestinal Barrier and Hematopoiesis
A preclinical study in Advanced Science showed that Faecalibaculum rodentium and its metabolite butyrate can alleviate ionizing radiation–induced damage in a mouse model by improving intestinal integrity and hematopoiesis. (DOI: https://doi.org/10.1002/advs.202509383). Diversity sequencing tracked changes in gut microbial composition under different radiation and genetic backgrounds, while metagenomics and metabolomics linked butyrate-producing pathways to protective effects on the gut barrier and hematopoietic recovery. This is a clear example of how community-level shifts and functional pathways are jointly resolved.
Crop Rotation Systems: "Altruistic" Rhizosphere Microbiomes for Soil-Borne Disease Management
In Plant Communications, researchers described an "altruistic rhizo-microbiome strategy" in garlic–chili crop rotation systems, where garlic root exudates drive the assembly of microbial communities that benefit subsequent crops but not garlic itself (DOI: https://doi.org/10.1016/j.xplc.2025.101502). Diversity sequencing tracked root-associated fungal and bacterial communities, while metagenomics and functional assays showed how garlic-derived diallyl disulfide (DADS) reshapes rhizosphere ROS stress and selects for Penicillium allii, which suppresses soil-borne pathogens in subsequent crops. The study demonstrates how integrated microbial diversity and metagenomic analysis can inform microbiome-based strategies for sustainable soil-borne disease management.
7. Designing Combined Microbial Diversity + Metagenomic Projects
Designing a joint microbial diversity and metagenomic sequencing strategy can feel complex, but a few practical patterns repeat across successful projects.
Typical Study Scenarios: Environmental, Agricultural, and Host-Associated Microbiota
Common use cases include:
- Environmental microbiomes: wastewater treatment, sediments, groundwater, extreme environments
- Agricultural and rhizosphere microbiomes: drought resilience, crop rotation, disease suppression
- Host-associated microbiota in research models: preclinical radiation injury, metabolic disorders, aging
In each scenario, you can start from a clearly framed biological question (e.g., "Which taxa and pathways drive methane yield?" or "Which root-associated microbes enhance drought resilience?") and then select sequencing layers accordingly.
Recommended Service Combinations on MicrobioSeq
CD Genomics' MicrobioSeq platform provides modular services that can be combined in one microbiome multi-omics study design:
 Figure 4. CD Genomics MicrobioSeq services combine 16S/18S/ITS microbial diversity profiling with shotgun metagenomics and integrated multi-omics study design to link community structure with functional genes and pathways.
Figure 4. CD Genomics MicrobioSeq services combine 16S/18S/ITS microbial diversity profiling with shotgun metagenomics and integrated multi-omics study design to link community structure with functional genes and pathways.
- Microbial Diversity Analysis – 16S/18S/ITS Sequencing for community structure
- Microbial Metagenomics and Metagenomic Shotgun Sequencing for functional genes and MAGs
- Metatranscriptomics for active pathways
- Microbial Functional Diversity Analysis and functional gene panels for targeted eco-functional questions
- eDNA Absolute Quantification strategies where relevant
Example Project Workflow and Practical Tips
A typical combined microbial diversity + metagenomics project on MicrobioSeq might look like:
 Figure 5. Typical workflow for a combined microbial diversity and metagenomics project, from pilot sampling and 16S/18S/ITS profiling to targeted metagenomics, MAG reconstruction, and optional absolute quantification and metabolomics.
Figure 5. Typical workflow for a combined microbial diversity and metagenomics project, from pilot sampling and 16S/18S/ITS profiling to targeted metagenomics, MAG reconstruction, and optional absolute quantification and metabolomics.
- Pilot sampling and metadata design
- Define treatments, gradients, and time points.
- Standardize sampling volumes and preservation methods.
- Microbial diversity sequencing (16S/18S/ITS)
- Profile all samples to understand community shifts.
- Use ordination and differential abundance to identify key contrasts.
- Select a subset for metagenomics / metatranscriptomics
- Choose representative and extreme samples, plus replicates.
- Plan sequencing depth and host DNA removal if needed.
- Metagenomic assembly, binning, and functional annotation
- Generate MAGs, pathway profiles, and functional gene tables.
- Integrate with diversity metrics and environmental or host data.
- Optional: absolute quantification and metabolomics
- Add qPCR/dPCR for total load and key functional genes.
- Include untargeted or targeted metabolomics to connect genes with metabolites.
From project experience, a few operational suggestions:
- Keep one data model from the start: define sample IDs, metadata fields, and directory structures early; this makes multi-omics integration and data traceability far easier.
- Plan for re-analysis: store raw reads, intermediate files, and analysis scripts; high-impact papers often revisit data for new questions.
- Align sequencing design with statistical analysis: consult with bioinformaticians or statisticians early to ensure power and replication are sufficient for the intended models.
All CD Genomics MicrobioSeq services are provided for research use only and are not intended for clinical diagnosis, treatment, or individual health assessments.
If you are planning a new microbiome project and are unsure how to balance 16S/18S/ITS amplicon sequencing, shotgun metagenomic sequencing, and additional layers such as metatranscriptomics or metabolomics, CD Genomics' MicrobioSeq team can help you design a fit-for-purpose, research-only workflow. By combining microbial diversity analysis, metagenomics, and optional absolute quantification, you can move from community-level profiles to strain-level functional insights in a single, integrated microbiome study.
8. FAQs: Combining Microbial Diversity and Metagenomic Sequencing
Q1. When is it better to combine 16S/18S/ITS amplicon sequencing and metagenomic sequencing instead of choosing just one method?
If you need both broad coverage across many samples and deep functional insight, a joint design is usually better. Use amplicon sequencing for high-throughput community profiling and to identify key contrasts, then add metagenomics on a carefully chosen subset to understand functions, MAGs, and pathways in detail.
Q2. How many samples should I send for shotgun metagenomic sequencing in a combined microbiome study?
There is no universal number, but a common pattern is full amplicon coverage across all samples, then metagenomics on 10–30 strategically selected samples that represent key states or transitions. The exact number depends on your budget, expected effect sizes, and the complexity of the microbiome.
Q3. Do I always need metatranscriptomics on top of metagenomics?
Not necessarily. Metatranscriptomics adds value when gene expression dynamics are central to your question—for example, in fast-changing bioreactors or stress-response models. If your primary goal is to map metabolic potential and MAGs, metagenomics alone may be sufficient. CD Genomics can help you decide whether the additional complexity of metatranscriptomics is justified for your research aims.
Q4. How can I ensure that my multi-omics microbiome data are reproducible and traceable?
Use consistent sample IDs, standardized protocols, and detailed metadata from the start. Store raw reads, intermediate outputs, and analysis scripts in an organized structure. When possible, deposit data in public repositories and cite DOIs for reference datasets, as done in the case studies above. CD Genomics can provide structured reports and data packages that support this level of traceability for microbiome research projects.
References
- Chetty, Ashwin, and Ran Blekhman. "Multi-Omic Approaches for Host-Microbiome Data Integration." Gut Microbes, vol. 16, no. 1, 2024, article 2297860.
- Kang, Jiamu, et al. "Integrated Multi-Omics Approaches to Understand Microbiome Assembly in Jiuqu, a Mixed-Culture Starter." Comprehensive Reviews in Food Science and Food Safety, vol. 21, no. 5, 2022, pp. 4076–4107.
- Kieser, Silas, et al. "ATLAS: A Snakemake Workflow for Assembly, Annotation, and Genomic Binning of Metagenome Sequence Data." BMC Bioinformatics, vol. 21, 2020, article 257.
- Liu, Yong-Xu, et al. "A Practical Guide to Amplicon and Metagenomic Analysis of Microbiome Data." Protein & Cell, vol. 12, no. 5, 2021, pp. 315–330.
- Rasmussen, Jacob Agerbo, et al. "A Multi-Omics Approach Unravels Metagenomic and Metabolic Alterations of a Probiotic and Synbiotic Additive in Rainbow Trout (Oncorhynchus mykiss)." Microbiome, vol. 10, 2022, article 21.






 "
width="400" height="200" loading="lazy"
alt="Beyond Relative Abundance: The Complete Guide to eDNA Absolute Quantification">
"
width="400" height="200" loading="lazy"
alt="Beyond Relative Abundance: The Complete Guide to eDNA Absolute Quantification">


