
 Sample Submission Guidelines
Sample Submission Guidelines
WGBS vs. EM-seq: Key Differences in Principles, Pros, Cons, and Applications
In the field of epigenetics, accurate analysis of DNA methylation patterns is very important, because DNA methylation plays a central role in gene expression regulation, development process and disease occurrence and development. Bisulfite sequencing technology is the cornerstone of this field, in which Whole Genome Bisulfite Sequencing (WGBS) has long been regarded as the gold standard, while the emerging enzyme-mediated methylation sequencing (EM-seq) is gradually emerging, which brings new opportunities and changes for DNA methylation research.
In this paper, WGBS and EM-seq are compared, and their differences in principle, advantages and disadvantages, sample applicability and applications in developmental biology, tumor research, botany and other fields are expounded.
Brief Introduction of WGBS and EM-seq
WGBS converts unmethylated cytosine into uracil by treating genomic DNA with bisulfite, while methylated cytosine remains unchanged, and then carries out full coverage methylation analysis on the treated DNA by high-throughput sequencing platform, which can accurately detect the methylation level of all single cytosine bases in the whole genome, and is widely used in cell differentiation, tissue development, animal and plant breeding, human health and disease research and other fields.
Em-seq is a new DNA methylation detection technology. It uses a variety of enzymes to convert unmodified cytosine into uracil on the premise of ensuring that methylated 5mC and hydroxymethylated 5hmC are still identified as C in sequencing, and then identified as thymine in subsequent amplification and sequencing. This method is suitable for tissues, cells, body fluids and other samples, especially for micro fragile DNA samples such as ctDNA. Only DNA samples as low as 10 ng can be used for genome-wide 5mC modified sequencing with single base accuracy.
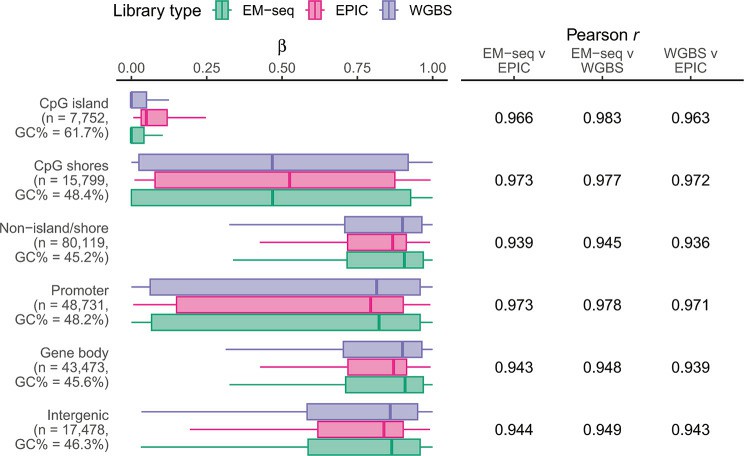 Methylation levels are well correlated in all biologically relevant genomic contexts across EM-seq and WGBS (Guanzon et al., 2024)
Methylation levels are well correlated in all biologically relevant genomic contexts across EM-seq and WGBS (Guanzon et al., 2024)
Services you may interested in
Learn More
Comparison of WGBS and EM-seq
Although the goals are the same, there are significant differences in their principles, experimental procedures, sample requirements and applications. An in-depth analysis of the differences between the two will help researchers make choice in technology selection, help them to exert their strength accurately in epigenetics research and unlock more life codes.
Different Principle of EM-seq and WGBS
WGBS technology is based on the fact that bisulfite can convert unmethylated cytosine into uracil, while methylated cytosine remains unchanged. First, genomic DNA is extracted and fragmented, and then incubated with bisulfite solution under specific conditions. After bisulfite treatment, unmethylated C was transformed into U, which was identified as thymine in the subsequent PCR amplification and sequencing, while methylated C still existed in the form of C. By analyzing the conversion rate from C to T in sequencing data, the methylation status of cytosine sites in DNA sequence can be accurately inferred, and the methylation map in the whole genome can be drawn.
EM-seq is a relatively new technology, which uses Ten-Eleven Translocation enzymes to oxidize 5mC to 5hmC, and then further to 5fC and 5caC. Then, uracil-DNA glycosylase (UDG) and deaminase (AID/APOBEC) converted unmethylated cytosine and oxidized 5fC and 5caC into uracil, while 5mC and 5hmC remained C in subsequent PCR amplification and sequencing because they were not transformed. By comparing the sequencing data before and after treatment, the DNA methylation site can be determined. This technique avoids some problems caused by bisulfite treatment, such as DNA degradation and fragmentation, and provides a mild and efficient alternative for DNA methylation analysis.
Differences in Advantages and Limitations
A. WGBS advantages
a) High resolution: WGBS can analyze the DNA methylation of the whole genome at single base resolution, and accurately identify the methylation status of each cytosine site, which enables researchers to deeply understand the details of methylation in gene regulatory regions and other functional elements, and provides an extremely accurate data basis for studying the relationship between methylation and gene expression.
a) Wide applicability: This technology is suitable for genomes of various species, whether it is model organisms such as mice, fruit flies, humans or other non-model organisms, as long as high-quality genomic DNA can be extracted, WGBS can be used for methylation research, which has a wide range of applications and universality.
B) EM-seq advantages
a) Less DNA damage: Compared with the strongly acidic bisulfite treatment in WGBS, the enzymatic reaction conditions of EM-seq are milder, and the damage to DNA is significantly reduced. This not only improves the success rate of processing low-quality or trace DNA samples, but also enables EM-seq to better preserve the integrity of DNA and obtain more reliable methylation data when it is difficult to obtain a large amount of DNA, such as precious clinical samples or ancient DNA samples.
b) Simplify library construction: EM-seq does not need complicated PCR amplification optimization steps after bisulfite conversion in the library construction process, which reduces the possibility of amplification bias. At the same time, due to the small DNA damage and low initial DNA consumption, a high-quality library can be constructed from ng-level DNA, which is of great significance for some research with limited sample size, greatly improving the experimental efficiency and reducing the cost.
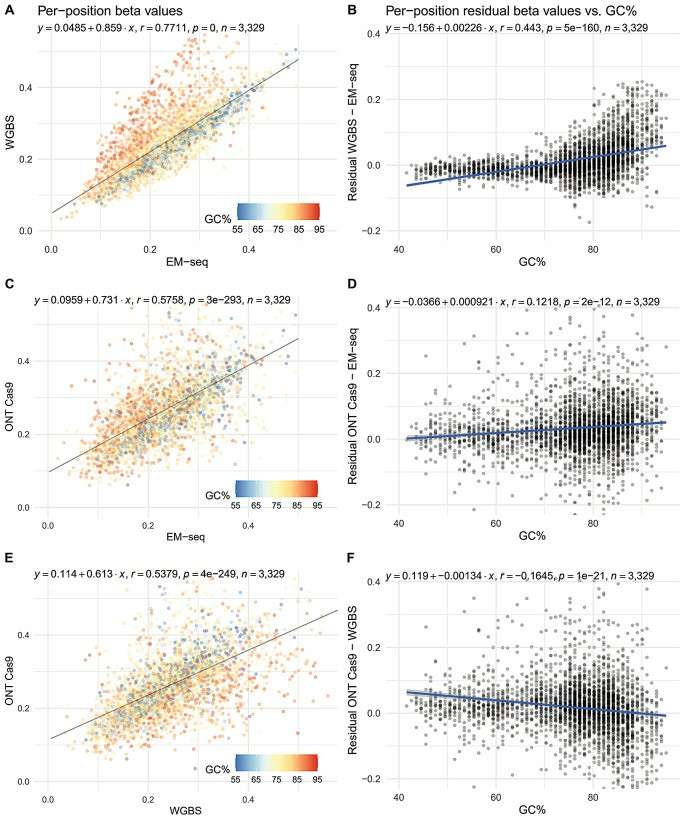 Pairwise comparisons of per-position methylation levels across the 45S locus (Guanzon et al., 2024)
Pairwise comparisons of per-position methylation levels across the 45S locus (Guanzon et al., 2024)
C. WGBS limitations
a) DNA degradation and fragmentation: The process of bisulfite treatment is severe, which will lead to a certain degree of DNA degradation and fragmentation. This may not only lose some DNA information, but also increase the difficulty of library construction. A large number of initial DNA is needed to ensure the success of the experiment, which may not meet the experimental requirements for some rare samples or low-abundance DNA sources.
b) Amplification bias: DNA treated with bisulfite is prone to amplification bias during PCR amplification, and some regions may have low amplification efficiency due to high GC content or other sequence characteristics, resulting in inaccurate acquisition of methylation information in these regions, which affects the integrity and accuracy of the whole genome methylation map.
D. EM-seq limitations
a) High cost: EM-seq technology requires the use of special enzymes and reagents, which are usually expensive. In addition, because the library construction process is complex, it needs to consume more samples and reagents, and the sequencing cost makes the overall experiment cost relatively high.
b) Complex data analysis: The data generated by EM-seq need to take into account its technical characteristics, such as digestion preference, base conversion efficiency and other factors. Compared with the traditional WGBS data, the analysis process of EM-seq data is more complicated, which requires special bioinformatics methods and software to process and interpret.
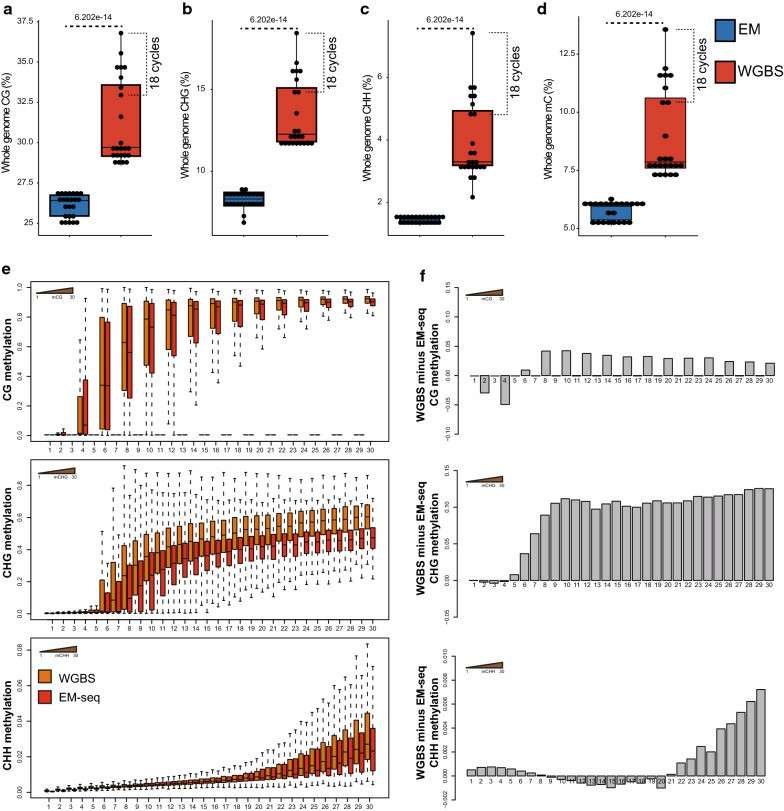 Comparison of methylation levels between EM-seq and WGBS (Feng et al., 2020)
Comparison of methylation levels between EM-seq and WGBS (Feng et al., 2020)
Sample Applicability Difference
WGBS: Due to the intense bisulfite treatment process, DNA will be degraded and fragmented to a certain extent, so WGBS usually needs a relatively large number of initial DNA samples. Generally speaking, for mammalian genome, the initial DNA amount needs to be at μg level. If the sample size is insufficient, it may not be possible to obtain enough high-quality sequencing data, which will affect the accuracy and reliability of the experimental results. Quality DNA samples are very important for WGBS. Impurities and degradation products in the sample may interfere with the bisulfite conversion reaction and the subsequent PCR amplification and sequencing process. Therefore, before the WGBS experiment, it is necessary to carry out strict quality inspection on the DNA sample, including concentration, purity (the ratio of OD260/280 should be between 1.8 and 2.0) and integrity (the integrity of DNA bands should be detected by agarose gel electrophoresis).
EM-seq: One of the obvious advantages of EM-seq technology is that it has little damage to DNA, so the amount of initial DNA required is low. This makes EM-seq have obvious advantages in dealing with precious samples or low-abundance DNA samples. For example, in ancient DNA research, single cell methylation analysis and clinical trace samples (such as circulating tumor DNA, ctDNA) detection, EM-seq can give full play to its low initial quantity and effectively obtain methylation information. Although EM-seq has little damage to DNA, the quality of samples will still affect the experimental results. However, compared with WGBS, the requirements of EM-seq on sample quality are relatively loose, but in order to ensure the best experimental effect, it is also recommended to use high-quality DNA samples for EM-seq experiments.
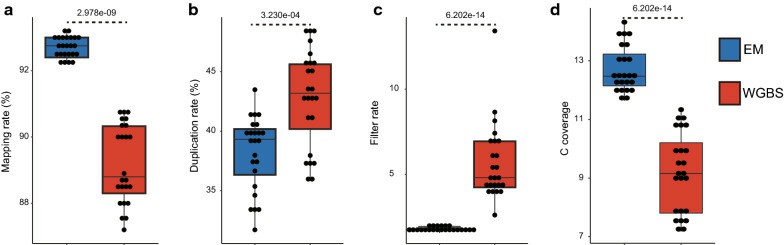 Quality comparison between EM-seq and WGBS (Feng et al., 2020)
Quality comparison between EM-seq and WGBS (Feng et al., 2020)
WGBS and EM-seq Various Applications in Research
DNA methylation plays a key role in many life processes such as biological growth and development, disease occurrence and so on. EM-seq and WGBS are the core technologies to detect DNA methylation. However, there are significant differences in their applications, and it is very important for researchers to understand these differences to select appropriate technologies and help research advance smoothly.
Developmental Biology Research
WGBS: During embryonic development, DNA methylation patterns show dynamic changes, which play a key role in cell differentiation and tissue and organ formation. With the advantage of single-base resolution, WGBS can accurately map the whole genome methylation map of embryonic cells at different development stages. However, because WGBS needs a lot of initial DNA, it is often challenging to obtain enough DNA for some early embryo samples or rare cell types (such as single cells in preimplantation embryos). In addition, bisulfite treatment may lead to DNA degradation, and there is a risk of sample loss for precious embryo samples.
EM-seq: The low starting amount of EM-seq makes it have unique advantages in some special sample studies of developmental biology. When studying embryo development at single cell level, the content of DNA in single cell is very small, which makes it difficult for traditional WGBS to meet the demand. However, EM-seq can successfully construct a methylation sequencing library from the DNA of single cell, revealing the methylation regulation mechanism at single cell level. At the same time, EM-seq has little damage to DNA, which is helpful to better preserve the original methylation information of DNA in embryo samples. EM-seq can provide more accurate methylation data and avoid misinterpretation of methylation information caused by DNA damage when studying some developmental processes sensitive to DNA damage, such as germ cell development.
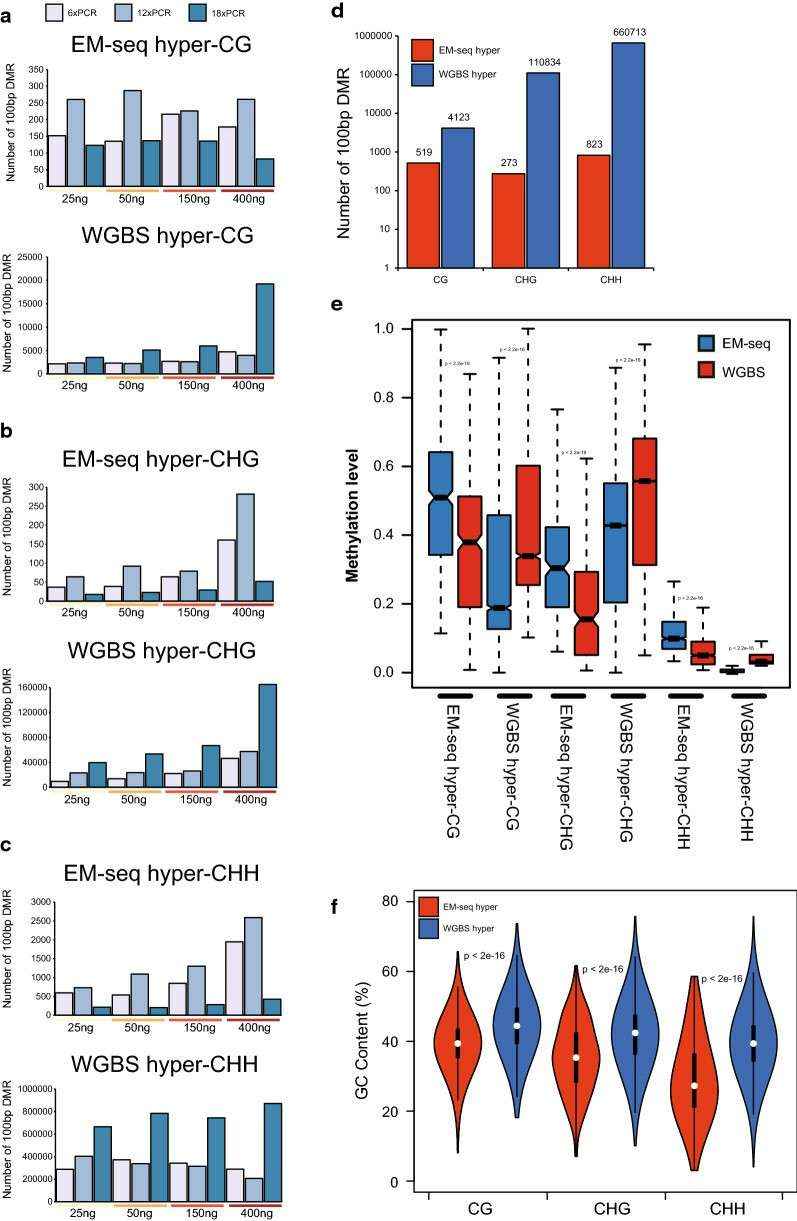 Differences in differentially methylated regions (DMRs) between EM-seq and WGBS (Feng et al., 2020)
Differences in differentially methylated regions (DMRs) between EM-seq and WGBS (Feng et al., 2020)
Tumor Research
WGBS: The occurrence and development of tumors are closely related to abnormal DNA methylation, including hypermethylation of tumor suppressor genes leading to gene silencing and hypomethylation of oncogenes promoting their expression. WGBS can comprehensively analyze the methylation differences between tumor tissues and normal tissues, and identify methylation markers related to tumor occurrence, development, metastasis and prognosis. However, WGBS also has some limitations in tumor research. Heterogeneity often exists in tumor tissues, and methylation patterns of different tumor cell subsets may be different. Bias in bisulfite treatment and PCR amplification may lead to the loss or misjudgment of some methylation information, which may affect the accurate evaluation of tumor heterogeneity. In addition, for some trace samples (such as biopsy samples) from tumor patients, WGBS may not be able to conduct effective analysis due to insufficient sample size.
EM-seq: The application of EM-seq in tumor research has attracted more and more attention, especially in the field of liquid biopsy. Liquid biopsy can realize early diagnosis, treatment monitoring and prognosis evaluation of tumors by detecting tumor markers (such as ctDNA) in body fluids such as blood. Because the content of ctDNA in blood is extremely low and easily degraded by various factors, it is difficult for traditional WGBS to analyze it effectively. EM-seq can accurately detect methylation information from a very small amount of ctDNA with its advantages of low initial amount and little damage to DNA. At the same time, EM-seq also has certain potential in analyzing tumor tissue heterogeneity. Because it can retain DNA information more completely, it may reflect the methylation difference of different cell subsets in tumor tissue more accurately, which is helpful to understand the heterogeneity of tumor and the evolution process of tumor cells.
Botany Research
WGBS: In the field of plant research, WGBS is widely used to analyze the methylation regulation mechanism during plant development, the response of plants to environmental stress and plant evolution. However, there are often a large number of repetitive sequences and high GC content regions in plant genomes, and WGBS is prone to amplification bias when dealing with these regions, which leads to inaccurate methylation information. In addition, plant samples may contain many impurities such as polysaccharides and polyphenols, which may interfere with the bisulfite conversion reaction and the subsequent experimental process and affect the reliability of the experimental results.
EM-seq: Studies show that EM-seq has unique advantages in the study of plant methylation. Because of its mild enzymatic reaction conditions and little damage to DNA, it can better deal with complex components in plant samples and reduce the interference of impurities on experiments. Compared with WGBS, EM-seq has higher accuracy and reliability in detecting DNA methylation, and is superior to WGBS in repeatability, mapping rate, repetition rate, background noise, average coverage and total cytosine coverage. In addition, EM-seq has also made important discoveries in detecting the dynamic methylation of plant genomes. Using EM-seq, researchers found a new type of genome methylation (GBM) in Arabidopsis thaliana, which is in a dynamic change of continuous addition and removal in all cells of plants, including germ cells. This dynamic GBM is evolutionarily conservative and related to enhanced gene expression plasticity.
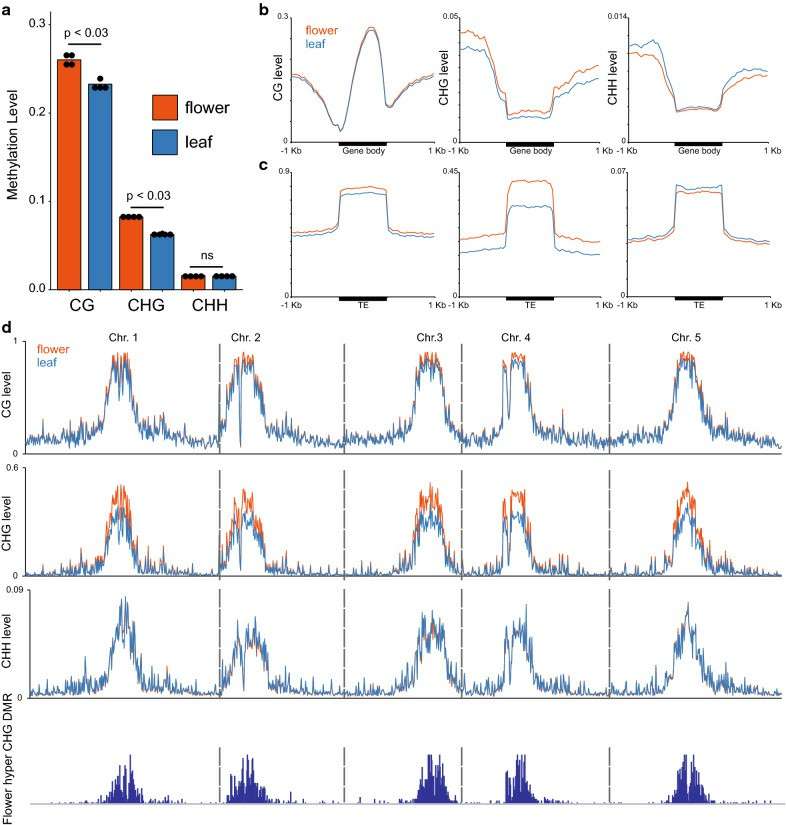 Methylation levels in Arabidopsis leaf and flower samples detected by EM-seq (Feng et al., 2020)
Methylation levels in Arabidopsis leaf and flower samples detected by EM-seq (Feng et al., 2020)
Research in Other Fields
WGBS: In microbial research, WGBS can be used to analyze the methylation patterns of microbial genomes such as bacteria and fungi, and understand the role of methylation in microbial gene expression regulation, virulence factor expression and adaptation to the environment. However, the structure and composition of microbial genome are quite different from those of eukaryotes, so WGBS may need to make some optimization according to its characteristics, such as adjusting bisulfite treatment conditions and PCR amplification parameters. In the study of ancient DNA, although WGBS can theoretically be used to analyze the DNA methylation pattern of ancient organisms, the application of WGBS in the study of ancient DNA is greatly limited because of the extremely low content of ancient DNA samples and the serious degradation of most of them, and the problems of DNA loss and fragmentation during bisulfite treatment.
EM-seq: EM-seq shows great potential in the study of ancient DNA. Because of its little damage to DNA and low initial requirement, it can be used for methylation analysis of extremely small and severely degraded ancient DNA samples. For example, when studying the methylation of DNA in ancient human teeth, EM-seq successfully detected the methylation information in ancient human genome, which provided a new perspective for studying the evolution, migration and disease evolution of ancient human beings. In the field of animal breeding, EM-seq can be used to screen methylation markers related to important economic traits of animals.
Conclusion
As two important DNA methylation sequencing technologies, EM-seq and WGBS have many differences in application. With its long-term accumulated experience and wide application, WGBS still plays an important role in some routine samples and research fields. However, its high requirements for sample size and quality, DNA damage and amplification bias limit its application in some special samples and complex research scenarios. In contrast, EM-seq, as a new technology, has shown great potential in the research of precious samples (such as ancient DNA and single cells), liquid biopsy and research fields that require high DNA integrity with its advantages of low initial amount, little damage to DNA and high data quality.
With the continuous development and improvement of technology, the two technologies may complement each other in different application scenarios in the future, providing more comprehensive and accurate technical support for DNA methylation research and helping us to deeply understand the epigenetic regulation mechanism in life process and the occurrence and development mechanism of related diseases. When choosing whether to use EM-seq or WGBS, researchers need to comprehensively consider many factors such as sample type, research purpose, data quality requirements and experimental cost, so as to choose the most suitable technical scheme for their own research.
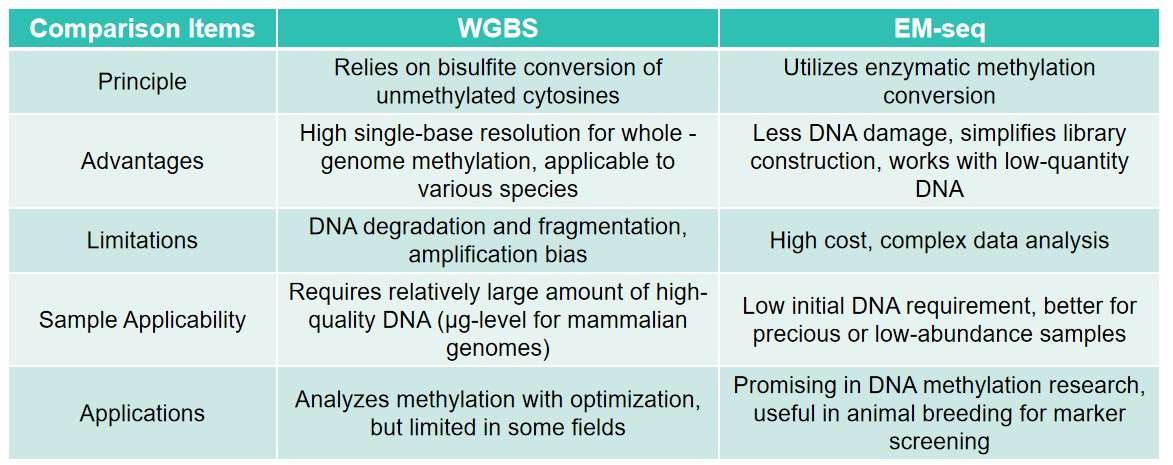 Summary of comparison between WGBS and EM-seq
Summary of comparison between WGBS and EM-seq
References:
- Guanzon D, Ross JP., et al. "Comparing methylation levels assayed in GC-rich regions with current and emerging methods." BMC Genomics. 2024 25(1):741 https://doi.org/10.1186/s12864-024-10605-7
- Feng S, Zhong Z., et al. "Efficient and accurate determination of genome-wide DNA methylation patterns in Arabidopsis thaliana with enzymatic methyl sequencing." Epigenetics Chromatin. 2020 13(1):42 https://doi.org/10.1186/s13072-020-00361-9
- Vaisvila R, Ponnaluri VKC., et al. "Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA." Genome Res. 2021 31(7):1280-1289 https://doi.org/10.1101/gr.266551.120
- Olova NN, Andrews S. "Whole Genome Methylation Sequencing via Enzymatic Conversion (EM-seq): Protocol, Data Processing, and Analysis." Methods Mol Biol. 2025 2866:73-98 https://doi.org/10.1007/978-1-0716-4192-7_5
- Ehrlich KC, Lacey M., et al. "Promoter-Adjacent DNA Hypermethylation Can Downmodulate Gene Expression: TBX15 in the Muscle Lineage." Epigenomes. 2022 Dec 6(4):43 https://doi.org/10.3390/epigenomes6040043
- Vaisvila R, Johnson SR., et al. "Discovery of cytosine deaminases enables base-resolution methylome mapping using a single enzyme." Mol Cell. 2024 84(5):854-866 https://doi.org/10.1016/j.molcel.2024.01.027