
 Sample Submission Guidelines
Sample Submission Guidelines
EM-seq: Principles, Workflow, Applications, and Challenges
In the vast field of biology science, epigenetic research is gradually becoming one of the core frontiers to explore the mysteries of life. Epigenetic modifications such as DNA methylation play an important role in regulating gene expression, cell differentiation and development process. Accurate and efficient detection of these epigenetic markers is very important for understanding the molecular mechanism of life phenomena. Under this background, the technology of enzyme-mediated methylation sequencing (EM-seq) came into being. With its unique technical principle and obvious advantages, it opened a new era for epigenetics research, attracted the attention of many researchers in recent years, and greatly promoted the development of this field.
EM-seq is a cutting-edge technology for accurate detection of DNA methylation level. In today's life science research field, it is very important to explore the regulation mechanism of gene expression, and DNA methylation, as one of the key epigenetic modifications, has attracted much attention. With its unique technical principle and obvious advantages, EM-seq is gradually becoming a powerful tool for researchers to deeply analyze the mystery of methylation.
This paper introduces the enzyme-mediated methylation sequencing technology EM-seq, including its principle, workflow, technical advantages and applications in biology science research and has broad application prospects.
Services you may interested in
Learn More
The Principle of EM-seq
The core mechanism of EM-seq technology is mainly based on the synergistic effect of Ten-Eleven Translocation (TET) and APOBEC enzyme. In the whole reaction process, TET enzyme, as the "leading factor of oxidation", carried out precise chemical modification on 5-methylcytosine (5mC) by virtue of its strong oxidation activity. Specifically, TET enzyme can gradually oxidize 5mC in an orderly manner, so that it can be transformed into 5-hydroxymethylcytosine (5hmC) and 5-aldocytosine (5fC) through multiple intermediate states, and finally 5-carboxycytosine (5caC) can be generated. Each step of oxidation reaction is highly accurate and orderly, which lays a key foundation for the subsequent detection process.
At the same time, APOBEC enzyme also plays an indispensable role. As a "deamination executor" of unmodified cytosine, APOBEC enzyme has high specific recognition ability for unmodified cytosine. Once the target is recognized, APOBEC enzyme quickly performs its deamination function and converts unmodified cytosine into uracil. It is worth noting that in the subsequent PCR amplification process, uracil will be accurately identified as thymine, which provides a key clue for distinguishing methylated and unmethylated sites. However, due to the relative stability of its own structure, 5mC and its oxidation products can resist the deamination of APOBEC enzyme and maintain the integrity of its own structure.
Based on the different action characteristics of these two enzymes, in the final sequencing results, the original methylation site remained as cytosine because it was not affected by APOBEC enzyme. However, the unmethylated site was transformed into thymine due to deamination of APOBEC enzyme and base recognition conversion during PCR amplification. Through this remarkable difference, researchers can effectively and accurately distinguish methylated sites from unmethylated sites in DNA sequences, which provides a strong technical support for further exploring the mechanism of DNA methylation in life process.
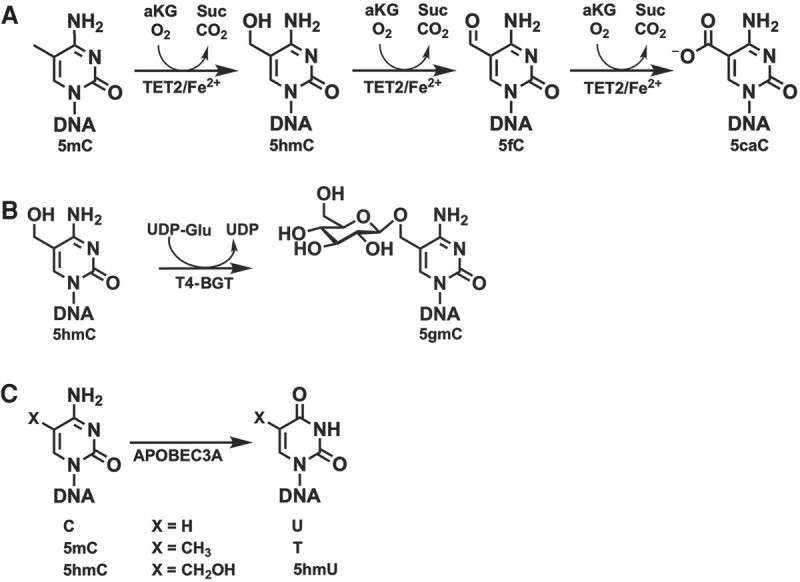 Enzymatic Activity of Enzymes Involved in Detection of 5mC and 5hmC (Vaisvila et al., 2021)
Enzymatic Activity of Enzymes Involved in Detection of 5mC and 5hmC (Vaisvila et al., 2021)
How Does EM-seq Work
As a new sequencing technology, EM-seq shows remarkable potential in accurately analyzing DNA methylation status. Its core lies in skillfully modifying DNA through enzymatic reaction, so as to convert methylation information into signals that can be recognized by high-throughput sequencing. This process abandons some complicated chemical treatment steps that may lead to DNA damage in traditional methods, greatly improves the accuracy and reliability of the experiment, and provides a new perspective and way for researchers to reveal methylation regulation mechanism in developmental biology, oncology and neuroscience.
DNA extraction: According to the source of the sample, choose the appropriate DNA extraction method. Blood samples can use commercial blood DNA extraction kits; Tissue samples need to be lysed first, and then DNA; is extracted by phenol-chloroform extraction or silica gel column adsorption. Cell line samples can be digested with cell lysate combined with protease K to extract DNA. The extracted DNA needs to be tested for purity and concentration to ensure that it meets the experimental requirements.
TET enzyme oxidation: A proper amount of DNA samples are mixed with TET enzyme and corresponding buffer, and incubated in PCR instrument. Generally, the reaction condition is incubation at 37°C for 1-2 hours, so that TET enzyme can oxidize 5mC to 5hmC and other subsequent treatable modified bases. This step makes methylated cytosine differ from unmethylated cytosine in chemical properties for subsequent differentiation.
Bisulfite treatment: The oxidized DNA sample is mixed with bisulfite reagent and reacted at a specific temperature and time. Generally, it is incubated at 50-60°C for 12-16 hours to convert unmethylated cytosine into uracil, while methylated cytosine remains unchanged. This step is the key step to distinguish methylated and unmethylated cytosine by EM-seq technology.
Library construction: The DNA library treated by EM-seq enzyme was amplified by PCR reaction to obtain a sufficient number of library molecules for subsequent sequencing. In the amplification process, primers were designed according to the linker sequence to ensure the effective amplification of library fragments.
High-throughput sequencing: The constructed library was loaded on the platform of high-throughput sequencing and sequenced according to the standard operating procedures of the sequencer. During the sequencing process, the instrument will sequence each DNA fragment in the library and generate a large number of original sequencing data, which are usually stored in FASTQ format.
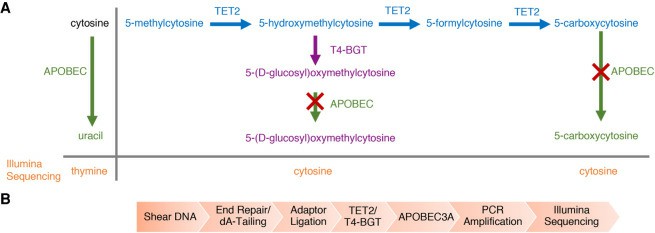 Enzymatic Methyl-seq mechanism of action and workflow (Vaisvila et al., 2021)
Enzymatic Methyl-seq mechanism of action and workflow (Vaisvila et al., 2021)
EM-seq Technological Advantages
High accuracy: Traditional bisulfite sequencing will degrade DNA a lot, resulting in information loss and affecting the accuracy of the results. However, EM-seq accurately converts unmethylated cytosine into uracil, while retaining methylated cytosine, which greatly reduces the errors caused by chemical treatment and can more truly reflect the methylation state of DNA.
Small sample demand: EM-seq has very low demand for the initial sample, and only a very small amount of DNA samples are needed to carry out the experiment. For those precious clinical samples with limited sample size, such as biopsy tissues and trace embryonic cells, EM-seq technology is undoubtedly an ideal choice, which effectively avoids the dilemma that methylation analysis cannot be carried out due to insufficient samples.
Simple operation: EM-seq is simpler in the experimental operation flow, and the bisulfite treatment steps are complicated, which requires long-term incubation and multiple purification. However, the enzyme transformation process of EM-seq is relatively simple, and the experimental period is greatly shortened, which not only improves the experimental efficiency, but also reduces the human error in the experimental process, making the experimental results more repeatable.
Good compatibility: EM-seq can be combined with a variety of downstream sequencing platforms, and it can be perfectly adapted to mainstream sequencing platforms such as Illumina, PacBio and Nanopore, which provides convenience for researchers to flexibly choose sequencing methods according to their own experimental needs and existing equipment conditions, and further broadens the application scope of this technology.
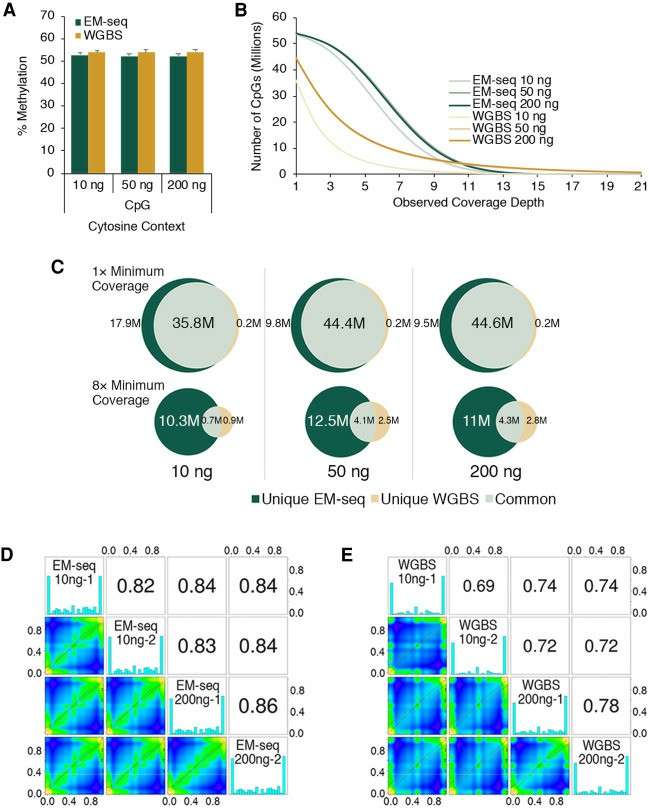 EM-seq has superior uniformity of GC coverage (Vaisvila et al., 2021)
EM-seq has superior uniformity of GC coverage (Vaisvila et al., 2021)
EM-seq Practical Application
As an innovative sequencing method, EM-Seq technology, with its unique enzymatic treatment strategy, has broken through the limitations of traditional technology and shown great application potential in many fields, and is gradually becoming a powerful tool for researchers to explore the mysteries of life, overcome medical problems and promote the development of related industries such as agriculture.
Disease Field
Cancer research: DNA methylation plays a key role in the occurrence and development of tumors. EM-seq technology can help researchers accurately find abnormal methylation sites related to cancer, thus providing a strong basis for early diagnosis, prognosis evaluation and development of new therapeutic targets of cancer. Through the comparative analysis of DNA methylation patterns between tumor tissues and normal tissues, it is expected to find some specific methylation markers for early screening of cancer.
Neuroscience research: The normal function of nervous system depends on precise gene expression regulation, and DNA methylation plays an important role in it. Using EM-seq technology, researchers can study methylation abnormalities in nervous system diseases (such as Alzheimer's disease, Parkinson's disease, etc.), explore the pathogenesis of these diseases, and open up new ways for finding potential treatments.
Developmental Biology
Embryo development research: In the process of embryo development, EM-Seq technology can be used to track the dynamic changes of DNA methylation of cells in different development stages and reveal the regulatory role of methylation in the process of cell fate determination, tissue and organ formation. By sequencing the methylation of early embryonic cells, we can understand the variation of methylation in key developmental events such as embryo implantation and gastrulation, which provides important information for studying the molecular mechanism of embryo development.
Cell differentiation research: This technology can analyze the methylation changes of different cell types during differentiation, and clarify the effects of methylation on cell-specific gene expression and cell function maintenance. Taking the differentiation of hematopoietic stem cells as an example, the specific methylation sites of different hematopoietic progenitor cells in the process of differentiation into various blood cells can be found by EM-Seq technology, so as to deeply understand the regulation mechanism of hematopoietic cell differentiation.
Botany Research
Research on plant growth and development: In the process of plant growth and development, such as seed germination, seedling growth, flowering and fruiting, EM-Seq technology can be used to study the regulation of DNA methylation on gene expression and reveal the mechanism of methylation in plant morphogenesis and organ development.
Study on plant stress resistance: DNA methylation will change dynamically when plants respond to biological stress (such as pests and diseases) and abiotic stress (such as drought, salinity and low temperature). EM-Seq technology can be used to analyze these changes, dig up methylation sites and genes related to stress resistance, and provide theoretical support for improving crop varieties and improving crop stress resistance.
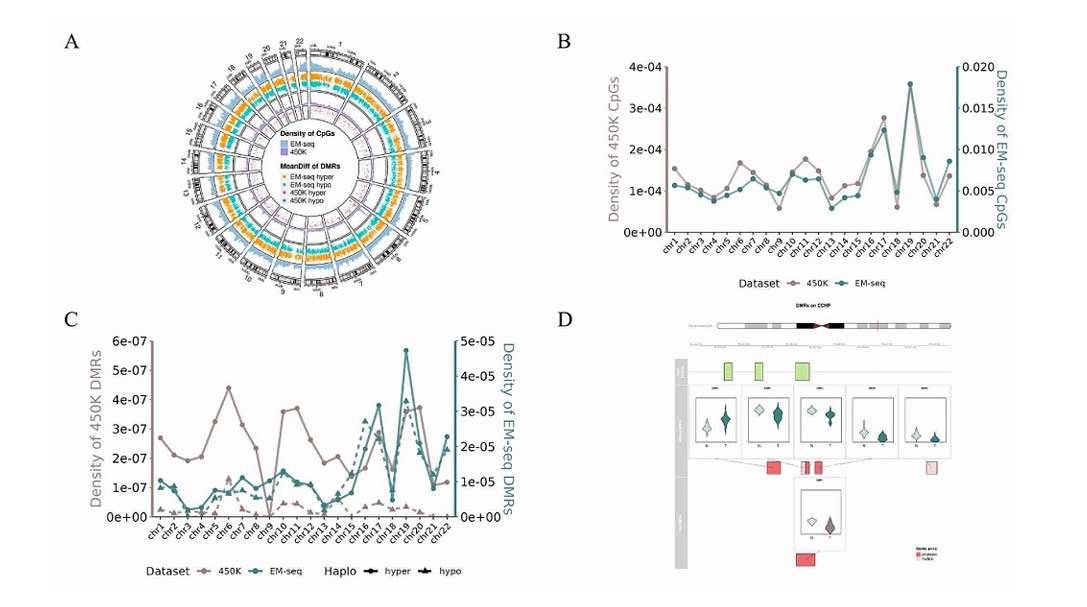 Distribution of DMRs across the whole genome (Gan et al., 2024)
Distribution of DMRs across the whole genome (Gan et al., 2024)
EM-seq Challenges and Solutions
As a new and potential technology, EM-seq has emerged in many research fields, but it also faces a series of thorny challenges in the practical application process. These challenges not only affect the accuracy and reliability of experimental results, but also limit the efficient application of this technology in a wider range of scenarios.
DNA extraction and fragmentation: There may be DNA loss or degradation during the extraction process, which will affect the subsequent experiments. It is difficult to control the uniformity of fragment size during fragmentation. If the fragment is too long or too short, the efficiency and accuracy of sequencing will be reduced. Use high-quality DNA extraction kit and optimized extraction methods, such as selecting lysis buffer suitable for sample type, and strictly controlling parameters such as temperature and time during extraction. In the fragmentation step, methods such as enzyme digestion or ultrasonic crushing can be used, and the best fragmentation conditions can be determined through experimental optimization, and fragments of appropriate size can be screened by agarose gel electrophoresis.
Library construction: The low efficiency of connecting connectors will lead to insufficient library yield. In addition, deviation may be introduced during PCR amplification, which will make some areas over-amplified or under-amplified, which will affect the accuracy and coverage of sequencing results. Optimize the reaction conditions of linker connection, such as adjusting the ratio of linker to DNA fragment, reaction temperature and time. Use high-quality ligase to improve ligation efficiency. In the PCR amplification process, the low deviation PCR amplification kit was used to optimize the PCR reaction conditions, such as annealing temperature and cycle times. At the same time, the molecular barcode can be used to mark DNA fragments to correct the PCR amplification deviation.
Data volume and computing resources: EM-Seq generates a huge amount of data, which requires high computing resources, including memory, storage and computing speed. The process of data processing and analysis is complex, which requires a lot of time and energy. Use a high-performance computing server or cloud computing platform to meet the needs of data processing and analysis. Adopt parallel computing, distributed computing and other technologies to improve the calculation efficiency. At the same time, optimize the data analysis process and use efficient algorithms and software tools to reduce the calculation time and resource consumption.
Sample heterogeneity: Biological samples often have cell heterogeneity, and the methylation level is different in different cell types or cell states, which will affect the accurate analysis of methylation patterns. If the research object is a specific cell type, cell sorting techniques, such as flow cytometry and laser capture microdissection, can be used to separate the target cell population first, and then analyze it by EM-Seq. Single cell EM-Seq technology can also be used to study methylation patterns directly at the single cell level, so as to better analyze the heterogeneity of samples.
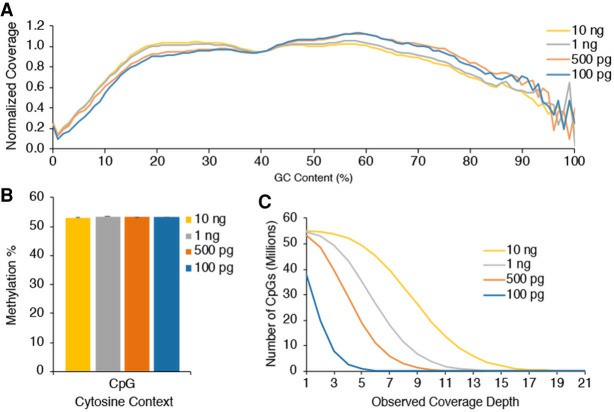 Low-DNA-input EM-seq libraries (Vaisvila et al., 2021)
Low-DNA-input EM-seq libraries (Vaisvila et al., 2021)
Conclusion
To sum up, EM-Seq has made great achievements in biomedicine, developmental biology, neuroscience and plant research by virtue of its unique advantages. It provides an accurate tool for analyzing DNA methylation patterns and helps to reveal the mystery of disease mechanism and development process. With the continuous optimization of technology, its application boundary will continue to expand, which is expected to break through more research bottlenecks, bring innovation to all fields of life science, shine more brightly in the future scientific research journey, and push human cognition of the nature of life to a new height.
References:
- Vaisvila R, Ponnaluri VKC., et al. "Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA." Genome Res. 2021 31(7):1280-1289 https://doi.org/10.1101/gr.266551.120
- Gan, J. Huang, M., et al. "Novel genome-wide DNA methylation profiling reveals distinct epigenetic landscape, prognostic model and cellular composition of early-stage lung adenocarcinoma." J Transl Med. 22 428 (2024) https://doi.org/10.1186/s12967-024-05146-2
- Bahado-Singh RO, Vishweswaraiah S., et al. "Placental DNA methylation changes and the early prediction of autism in full-term newborns." PLoS One. 2021 16(7):e0253340 https://doi.org/10.1371/journal.pone.0253340
- Černoša I, Trincado-Alonso F., et al. "Use of Enzymatically Converted Cell-Free DNA (cfDNA) Data for Copy Number Variation-Linked Fragmentation Analysis Allows for Early Colorectal Cancer Detection." Int J Mol Sci. 2024 25(6):3502 https://doi.org/10.3390/ijms25063502
- Olkhov-Mitsel E, Bapat B. "Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers." Cancer Med. 2012 1(2):237-60 https://doi.org/10.1002/cam4.22
- Han YL. "Advanced Research on DNA methylation testing in screening fetuses for autism spectrum disorder." Cell. Mol. Biomed. Rep. 2025, 5(1): 1-12 https://doi.org/10.55705/cmbr.2025.449757.1243