
 Sample Submission Guidelines
Sample Submission Guidelines
A Step-by-Step Guide to EM-Seq: From DNA Extraction to Methylation Analysis
Enzymatic Methyl-seq (EM-seq), as an innovative and efficient technology, is gradually emerging in the forefront of genomics research, providing a powerful tool for in-depth analysis of the mystery of DNA methylation. The process of EM-seq is precise and rigorous, which integrates a series of advanced experimental operations and scientific principles. From the initial processing of samples to the accurate acquisition of final data, every step plays a key role in accurately revealing DNA methylation patterns.
In this paper, EM-seq workflow is introduced in detail, including sample preparation, chemical modification, library construction, sequencing steps and data analysis, showing its important role and application potential in biology science research.
 A schematic of the EM-seq workflow (Olova et al., 2023)
A schematic of the EM-seq workflow (Olova et al., 2023)
EM-seq Sample Preparation
In the EM-seq experiment, sample preparation is a very critical initial step, and its quality is directly related to the accuracy and reliability of subsequent experimental results. From obtaining biological samples to processing them into samples suitable for EM-seq, every step needs to strictly follow the standard process and control many details to ensure that the DNA methylation information in the samples can be completely and accurately retained and analyzed.
Sample types and sources: There are various types of samples that can be used in the EM-Seq process. Cell line is one of the commonly used samples. Because of its relatively homogeneous characteristics, it is very convenient to study the epigenetic characteristics of specific cell types. When studying the epigenetic regulation mechanism of immune cells to understand the immune response process, the corresponding immune cell lines can be selected. Tissue samples cover a wide range, and tumor tissue samples are very important in the epigenetic study of cancer. By analyzing epigenetic modifications such as DNA methylation in tumor tissues, we can reveal the molecular mechanism of tumor occurrence and development, and provide key clues for early diagnosis, prognosis judgment and discovery of therapeutic targets of cancer.
Precautions for sample collection and preservation: All kinds of sample collection should follow the specific best method and opportunity. For tumor tissue samples, they should be collected as soon as possible after surgical resection to avoid the epigenetic modification of tumor cells due to ischemia and other factors. It is generally recommended to complete the collection within 30 minutes after in vitro. When collecting, ensure that the collection site is representative and try to avoid mixing with normal tissues. Tissue samples can be quickly frozen in liquid nitrogen after collection to maintain their epigenetic modification in vivo. Blood samples are usually collected by intravenous blood collection, and blood collection tubes containing anticoagulants are used. After blood collection, they should be gently inverted and mixed evenly in time to prevent blood coagulation.
Nucleic acid extraction: According to different sample types, appropriate nucleic acid extraction methods should be selected. For cell line samples, silica gel column adsorption method is commonly used, which is simple and quick to operate, can complete nucleic acid extraction in a short time, and is suitable for Qualcomm's quantity experiment. However, this method may have a certain amount of nucleic acid loss during elution. Phenol-chloroform method is often used to extract nucleic acid from tissue samples. The nucleic acid extracted by this method has high purity and can effectively remove impurities such as protein. However, the operation steps are complicated, and toxic reagents such as phenol and chloroform are needed, and the operating skills of experimenters are required.
Quality control: Key indicators include concentration, purity and integrity. The concentration is detected by a spectrophotometer. After the nucleic acid sample is diluted to a suitable multiple, it is put into a spectrophotometer to detect the absorbance at the wavelength of 260nm, and the nucleic acid concentration is calculated according to Lambert-Beer law. The purity is measured by the ratio of A260/A280 and A260/A230. The ratio of A260/A280 of pure DNA samples should be between 1.8 and 2.0. If the ratio is lower than 1.8, there may be protein pollution. If the ratio is higher than 2.0, there may be RNA pollution. The ratio of A260/A230 should be between 2.0 and 2.2. If the ratio is too low, there may be impurities such as polysaccharides and polyphenols. The integrity of nucleic acid was judged by observing the integrity of bands by agarose gel electrophoresis. After the nucleic acid sample is mixed with the loading buffer, it is added into the loading hole of agarose gel, and electrophoresis is carried out at a suitable voltage. If the bands are vague and diffuse, it indicates that the integrity of the nucleic acid is poor and may have been degraded, which will affect the subsequent EM-Seq experimental results.
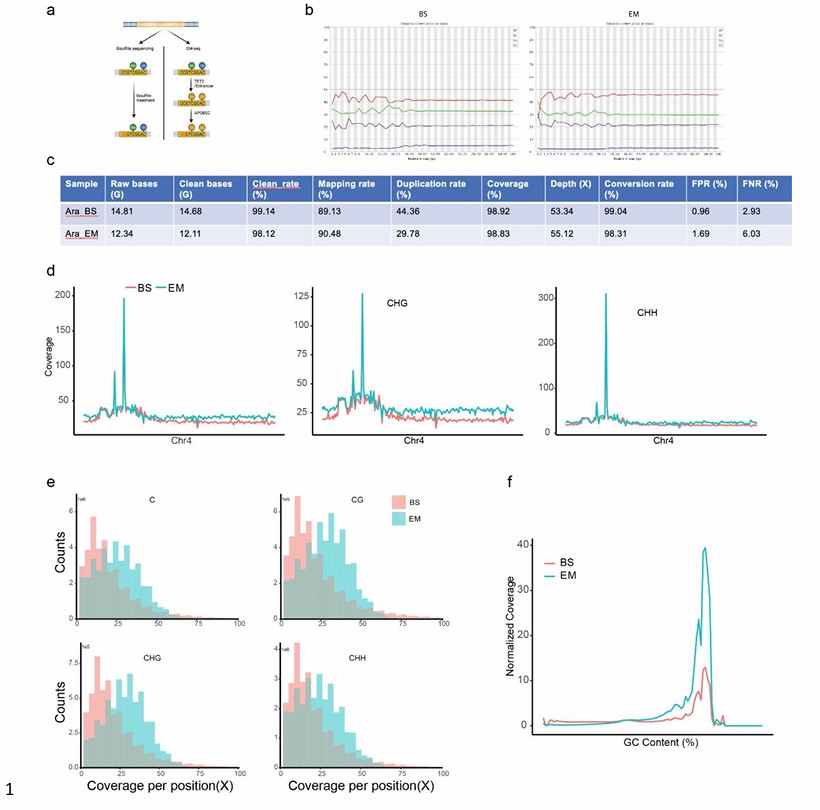 The quality control of the EM-seq and BS-seq (Wang et al., 2022)
The quality control of the EM-seq and BS-seq (Wang et al., 2022)
Services you may interested in
Learn More
Chemical Modification in EM-seq
The chemical modification process of EM-seq is not a direct chemical change of DNA bases in the traditional sense, but a series of well-designed enzymatic reactions to achieve efficient labeling and accurate detection of specific methylation sites. This chemical modification method has opened up a brand-new and potential path for comprehensively and accurately revealing the mystery of DNA methylation group, which has greatly promoted the development of related research.
Modification Reagent and Reaction
In the EM-Seq process, the key chemical modification reagents are β-glucosyltransferase (βGT) and bisulfite. βGT is an enzyme that can add glucose groups to 5-methylcytosine (5mC). Its chemical essence is protein, which is formed by folding a specific amino acid sequence. In the reaction, 5-methylcytosine -β-D-glucoside (5mC-β-D-glucopyranoside) was formed by adding a β-glucosyl group to the 5th carbon atom of 5mC by enzymatic reaction, which changed the chemical structure of 5mC. Its function is to label 5mC and distinguish it from unmodified cytosine, which lays a foundation for subsequent bisulfite treatment.
Bisulphite plays an extremely important role in EM-Seq. The chemical structure of bisulfite is HSO, which has strong reducibility. Under acidic conditions, bisulfite can deaminate with unmodified cytosine. The specific reaction mechanism is that bisulfite ion (HSO) first reacts with the amino group at specific position of cytosine to form an intermediate product. Subsequently, under certain conditions, the intermediate product undergoes deamination reaction, and the amino group is replaced by hydroxyl group, which is finally converted into uracil. However, methylation-modified cytosine has no effect on methylation-modified cytosine because of the existence of methyl group, which prevents the addition reaction between bisulfite and amino group at specific position.
Optimization of Reaction Conditions
During the modification reaction, temperature and time parameters have significant effects on the reaction efficiency and specificity. The results show that the reaction rate of bisulfite with unmodified cytosine is slow at lower temperature. Too high a temperature can also cause problems. When the temperature exceeds 60℃, although the reaction speed is extremely fast, there will be over-reaction. Overreaction is characterized by deamination of not only unmodified cytosine, but also partially methylated cytosine, and the probability of DNA strand breakage increases, which will seriously affect the accuracy of subsequent sequencing results. The concentration of modifying agent is also very important to the reaction effect. When the concentration of bisulfite is too low, the reaction of unmodified cytosine is incomplete, and it is impossible to label all unmodified bases effectively.
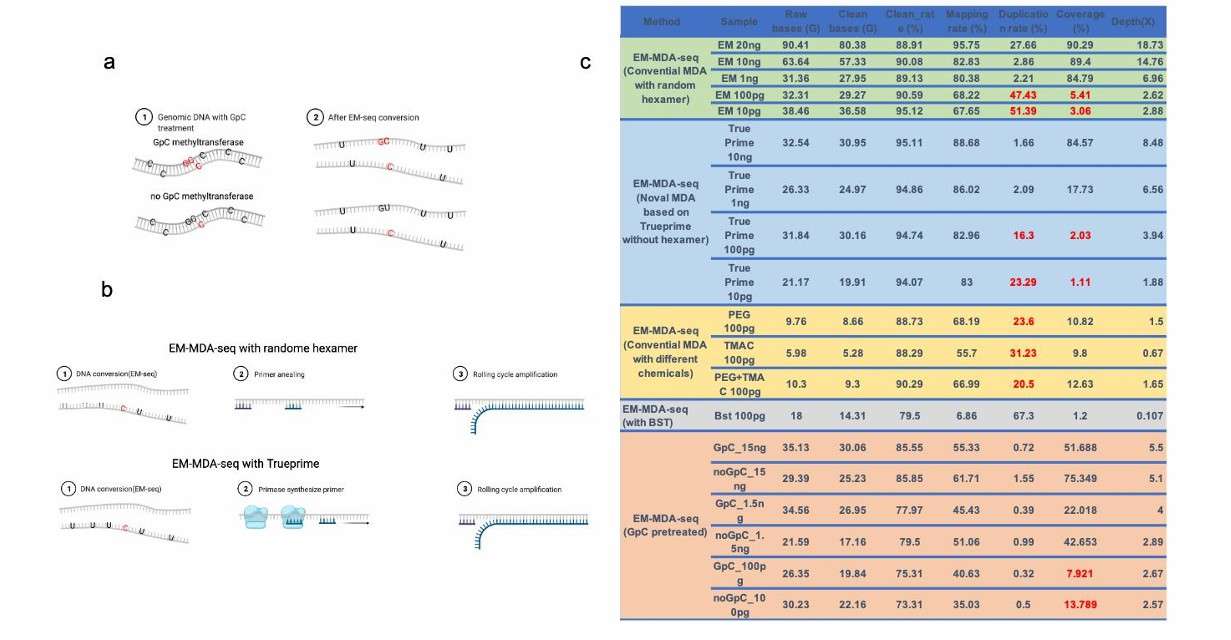 The MDA amplification on the EM-seq converted genome (Wang et al., 2022)
The MDA amplification on the EM-seq converted genome (Wang et al., 2022)
EM-seq Library Construction
In the construction stage of EM-seq library, fragmentation and linker ligation are crucial links, which directly affect the quality of the library and the accuracy and reliability of subsequent sequencing results.
Fragmentation Treatment
Physical fragmentation method: Ultrasonic fragmentation is to realize the fragmentation of nucleic acid molecules by using the mechanical effect of ultrasonic waves. When ultrasonic wave acts on nucleic acid solution, it will produce high-frequency vibration, which will form tiny bubbles in the solution. These bubbles expand and contract rapidly under the action of ultrasonic waves, and finally burst, resulting in strong shear force. Nucleic acid molecules are broken into fragments of appropriate size under the action of this shear force.
Enzymatic digestion: Enzymatic digestion is based on the recognition and cleavage of specific DNA sequences by restriction enzymes. Restriction endonucleases can recognize specific nucleotide sequences in double-stranded DNA molecules, which usually have palindrome structure. Once the target sequence is recognized, the restriction endonuclease will double-strand DNA at a specific site. Different restriction enzymes recognize different sequences, so appropriate restriction enzymes can be selected as needed to obtain DNA fragments with expected length range.
Connection Method
Universal primer binding site: This part of the sequence provides binding sites for universal primers in the subsequent PCR amplification, so that all fragments in the library can be effectively amplified in the PCR reaction. In the sequencing process, the sequencing primer is combined with this part of the sequence of the linker, thus guiding the sequencing reaction and ensuring that the sequence information of the nucleic acid fragment can be accurately read.
Selection of ligase: Different types of ligase have different ligation efficiency and substrate preference. T4 DNA ligase is a common ligase, which can efficiently connect sticky end and blunt end. In the ligation reaction, selecting T4 DNA ligase with high activity and good stability can improve the ligation efficiency.
Reaction temperature and time: The reaction temperature and time have significant influence on the connection effect. Generally speaking, a lower temperature (e.g. 16℃) is beneficial to stabilize the reaction system and improve the accuracy of the connection, but the reaction time needs to be relatively long (e.g. overnight connection); Higher temperature (e.g. 25℃) leads to faster reaction speed, but it may lead to lower connection specificity. Through experimental comparison, it is found that the quality and yield of the connected products are good when the temperature is 16℃ for 12 hours.
Molar ratio of linker to nucleic acid fragment: Proper molar ratio is the key to ensure the efficiency of ligation. When the molar ratio of linker to nucleic acid fragment is too high, it is easy to produce by-products such as linker dimer; If the molar ratio is too low, the connection efficiency is low, and some nucleic acid fragments in the library may not be connected to the linker. The optimization experiment showed that when the molar ratio of linker to nucleic acid fragment was 5:1, the connection effect was the best.
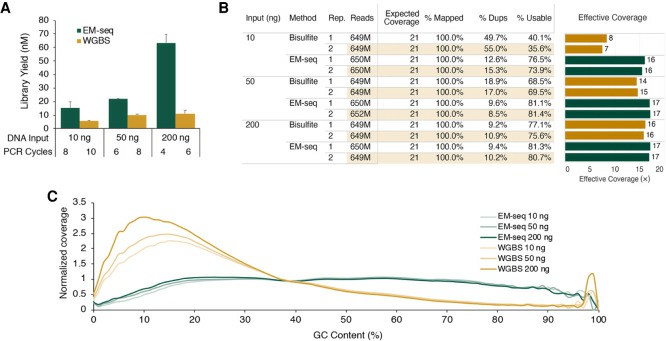 NA12878 EM-seq libraries (Vaisvila et al., 2021)
NA12878 EM-seq libraries (Vaisvila et al., 2021)
EM-seq Specific Sequencing Steps
After careful preparation such as enzymatic modification of samples and library construction, the sequencing stage is like a keen information catcher. With the advanced sequencing platform, DNA fragments with methylation markers are accurately read. Through a series of complex and orderly biochemical reactions and signal conversion, the base sequence and the corresponding methylation state are presented in digital form, which lays a solid foundation for the subsequent comprehensive and in-depth analysis of DNA methylation patterns and thus unlocks the mystery of its regulation in biological processes.
Library loading: When sequencing the EM-Seq library on the selected sequencing platform, the prepared library is first loaded into the sequencing chip or reaction cell. Taking Illumina platform as an example, the library is loaded through a special flow tank, and oligonucleotide sequences complementary to library connectors are fixed on the surface of the flow tank, and library fragments can be specifically bound to the surface of the flow tank to prepare for the subsequent sequencing reaction.
Sequencing reaction cycle: For the sequencing technology while synthesizing on Illumina platform, in the sequencing reaction cycle, four dNTP (deoxynucleotide) with different fluorescent labels are added into the reaction system in turn. DNA polymerase adds dNTP to the primer extension chain, and every time one base is added, it identifies the base type according to its fluorescence signal and records it. After many cycles, the whole library fragment was sequenced.
Data acquisition: With the sequencing reaction, the instrument collects fluorescence signals in real time and converts them into corresponding base sequence information. Sequencing data is finally produced in FASTQ format file.
Evaluation indicators and methods: The original sequencing data were preliminarily evaluated by FastQC software. In terms of base mass distribution, FastQC draws a mass distribution map by calculating the average mass fraction of bases at each position.
Quality filtering standard: Set quality filtering standard according to the evaluation result. Generally, bases with a base mass fraction of less than 20 (corresponding error rate is about 1%) are regarded as low-quality bases. When the proportion of low-quality bases in a sequence exceeds a certain threshold (such as 20%), the sequence is regarded as low-quality and removed. For linker contaminated sequences, sequences containing linkers are identified and removed by comparing known linker sequences.
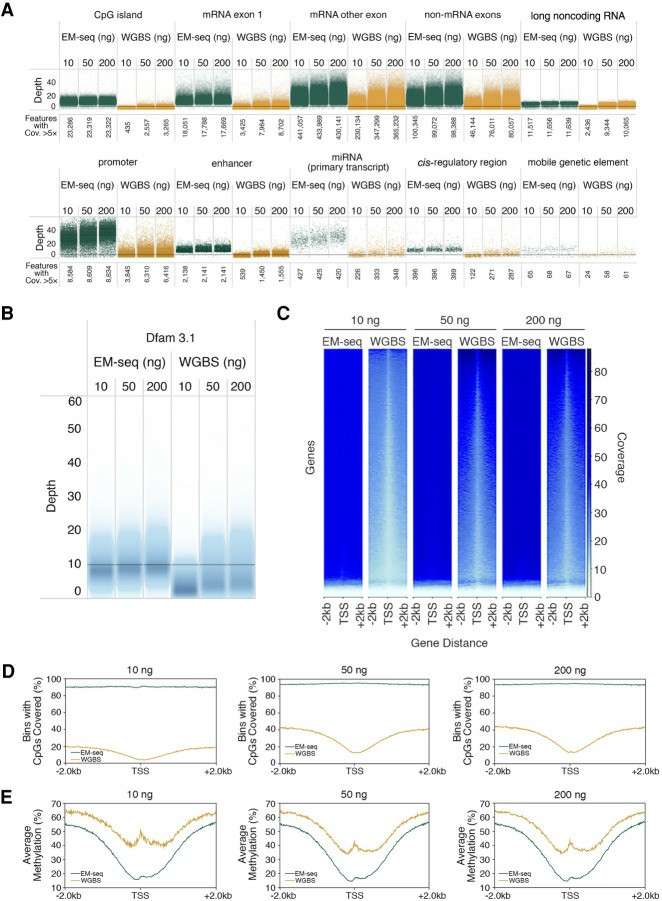 Cytosine methylation at key genomic features (Vaisvila et al., 2021)
Cytosine methylation at key genomic features (Vaisvila et al., 2021)
How to Analyse EM-seq Data
Through the deep excavation and analysis of complex data, we can not only understand the dynamic changes of DNA methylation in the process of cell differentiation and development, but also find methylation characteristics closely related to the occurrence and development of diseases in the field of disease research, such as cancer and neurodegenerative diseases, thus providing solid data support and theoretical basis for early diagnosis of diseases and formulation of precise treatment strategies.
Modification site identification: The algorithm principle of identifying epigenetic modification sites in EM-Seq data is based on comparing the characteristics of nucleic acid sequences before and after modification in sequencing data. In EM-Seq, methylation modification will lead to the signal characteristics of nucleic acid sequences different from those of unmodified sequences after specific treatment. Through the analysis of sequencing data, the methylation status of each locus was judged by statistical method and machine learning model.
Distribution and pattern analysis of modification sites: different genomic regions will be considered when the distribution of identified epigenetic modification sites on the genome is statistically analyzed. In the gene region, it includes promoter, exon and intron. The methylation status of promoter region is usually closely related to gene expression regulation, and hypermethylation often inhibits gene transcription. By counting the distribution of methylation sites in the promoter region, we can find out which gene promoters may be regulated by methylation. In exon and intron regions, methylation distribution also has its own characteristics, which may affect the splicing process of mRNA.
Integration analysis with other omics data: By integrating EM-Seq data with other omics data, we can fully understand the mechanism of epigenetic modification in biological process. Taking the integration with transcriptome data as an example, through the correlation analysis of the changes of methylation sites and gene expression levels, we can find the key genes that may be regulated by epigenetics. Generally, the expression level of genes with high methylation in the promoter region is often low; However, the expression level of hypomethylated genes may be higher.
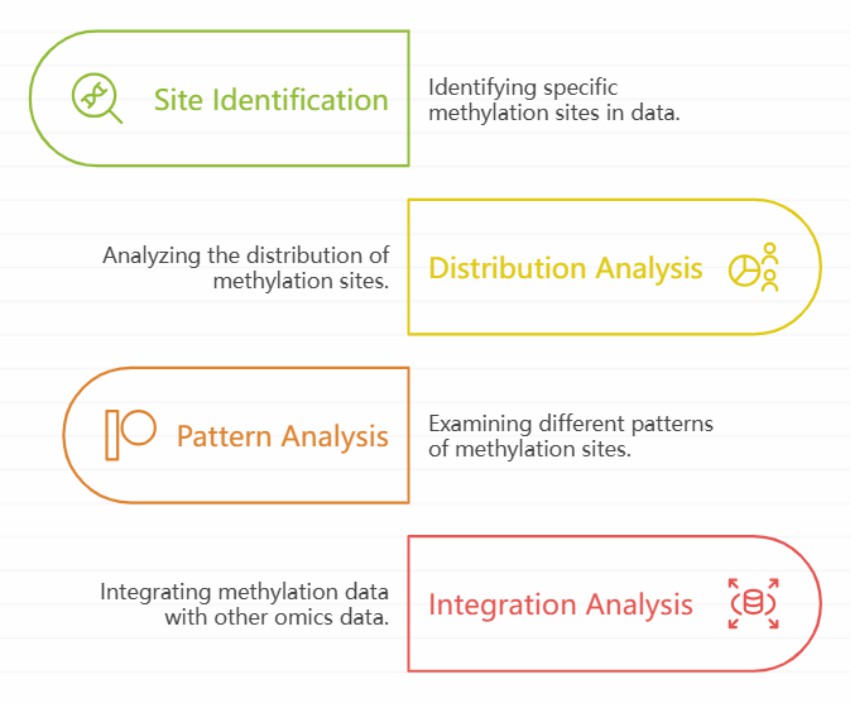 EM-seq biological analysis
EM-seq biological analysis
Conclusion
To sum up, the EM-seq process starts with sample DNA extraction, and goes through a series of precise steps such as TET enzyme treatment, terminal repair and linker connection, PCR amplification, etc., and finally obtains a sequencing library that can be used for in-depth analysis. With its high efficiency and accuracy, this process can accurately detect DNA methylation modification sites, provide a powerful tool for researchers to deeply explore biological issues such as gene expression regulation and disease occurrence and development mechanism, show great application potential in the field of life science research, and constantly push related research to a new height.
References:
- Berthold A, Lloyd VK. "Changes in the Transcriptome and Long Non-Coding RNAs but Not the Methylome Occur in Human Cells Exposed to Borrelia burgdorferi." Genes (Basel). 2024 15(8):1010 https://doi.org/10.3390/genes15081010
- Vaisvila R, Ponnaluri VKC., et al. "Enzymatic methyl sequencing detects DNA methylation at single-base resolution from picograms of DNA." Genome Res. 2021 31(7):1280-1289 https://doi.org/10.1101/gr.266551.120
- Feng S, Zhong Z., et al. "Efficient and accurate determination of genome-wide DNA methylation patterns in Arabidopsis thaliana with enzymatic methyl sequencing." Epigenetics Chromatin. 2020 13(1):42 https://doi.org/10.1186/s13072-020-00361-9
- Bouzeraa L, Martin H., et al. "Decoding epigenetic markers: implications of traits and genes through DNA methylation in resilience and susceptibility to mastitis in dairy cows." Epigenetics. 2024 19(1):2391602 https://doi.org/10.1080/15592294.2024.2391602
- Wang J, Fang YT., et al. "High coverage of single cell genomes by T7-assisted enzymatic methyl-sequencing." bioRxiv 2022: 25 https://doi.org/10.1101/2022.02.23.481567
- Williams CJ, Dai D., et al. "Dynamic DNA methylation turnover in gene bodies is associated with enhanced gene expression plasticity in plants." Genome Biol. 2023 24(1):227 https://doi.org/10.1186/s13059-023-03059-9