
 Sample Submission Guidelines
Sample Submission Guidelines
Targeted Bisulfite Sequencing
As a leading provider of NGS services and a partner of Illumina, CD Genomics is dedicated to providing a portfolio of solutions for epigenetic studies. Characterized by small sample size requirements, high-resolution, efficiency and economy, targeted bisulfite sequencing serves as a viable tool for methylation status analysis of targeted genomic regions. With over 10 years of professional experience, we can totally meet your project requirements and budgets in the exploration of methylome.
The Introduction of Targeted Bisulfite Sequencing
DNA methylation, happens most commonly at cytosines in the CpG sites, plays an important role in gene expression and regulation. The ability to detect and evaluate DNA methylation precisely and efficiently contributes to improving our understanding of DNA methylation in development and disease. While whole genome bisulfite sequencing identifies methylated cytosines on a whole-genome scale, targeted bisulfite sequencing is an accurate, efficient and economical technology for DNA methylation analysis of target regions, which may include a hybridization-based step on platforms containing pre-designed oligos that capture the CpG islands, gene promoters and other significant methylated regions, or a PCR-based step to amplify multiple bisulfite-converted DNA regions in a single reaction. There are commercially designed capture libraries available with a range of epigenetic features that cover about 12% to 24% or so of all genome CpGs. In addition, specific primers are designed to capture the region of interest and evaluate site-specific DNA methylation changes.
Advantages of Targeted Bisulfite Sequencing
- Single-base resolution DNA methylation patterning of selective regions.
- Enhanced accuracy and sensitivity while reducing your overall cost.
- Allowing for better determination of both SNPs and methylation status events.
- Providing a better understanding of development and disease.
Application of Targeted Bisulfite Sequencing
- Gene Regulation Mechanism Research
- Methylation Marker Screening
- Methylation Validation
- Animal and Plant Breeding Research
- Clinical Large-Scale Multi-Site Methylation Studies
Targeted Bisulfite Sequencing Workflow
Our highly experienced expert team and strict quality control following every procedure ensure the comprehensive and accurate results. In the process of targeted bisulfite sequencing, the genomic DNA is bisulfite converted and multiplex amplified using specific designed and validated primers or probes, the amplicons are pooled through barcoding and adapterization. The amplicon libraries are then subjected to sequencing with Illumina HiSeq platforms.

Service Specifications
Sample requirements and preparation
|
|
 |
Sequencing
|
 |
Data Analysis
We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
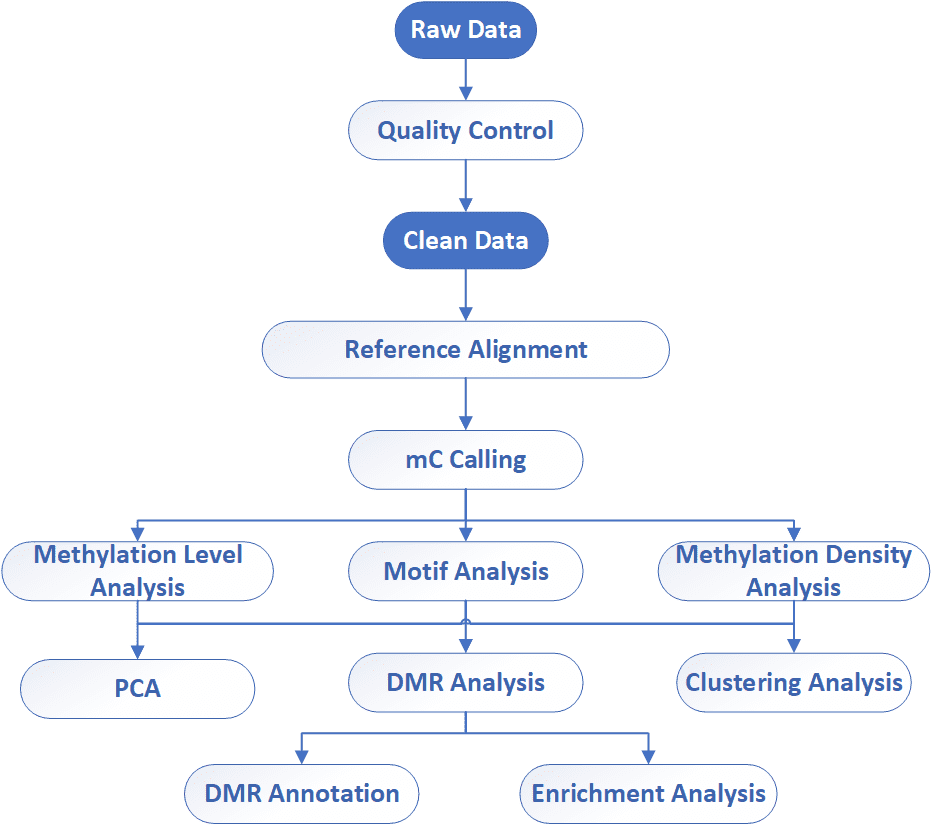
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in Targeted Bisulfite Sequencing for your writing (customization)
CD Genomics provides high-quality data and integrated bioinformatics analyses services for targeted bisulfite sequencing including bisulfite conversion, primer/probe design and validation, hybridization/PCR amplification, library preparation, DNA sequencing and data analysis. CD Genomics uses both commercially designed capture arrays and custom-designed capture libraries to fully meet your research objectives. Please feel free to contact us for more information. We look forward to cooperating with you in the near future.
Reference:
- Ziller M. J. et al. Targeted bisulfite sequencing of the dynamic DNA methylome. Epigenetics & chromatin, 2016, 9(1): 55.
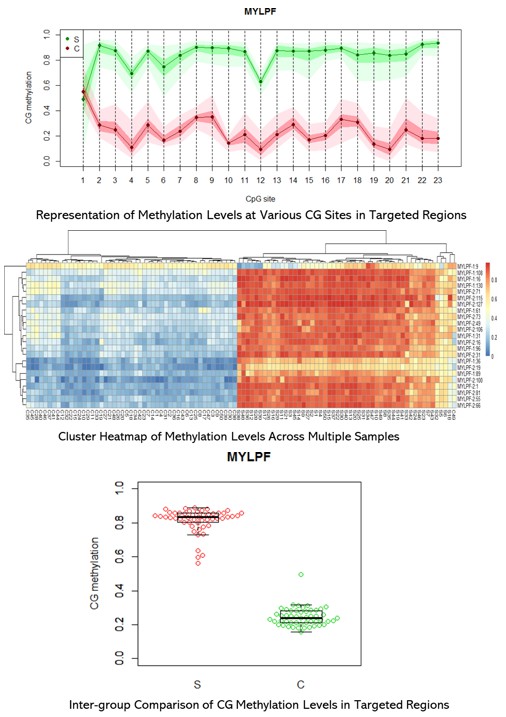
Demo Results
Targeted Bisulfite Seq FAQs
1. When you should choose targeted bisulfite sequencing?
The technology of targeted bisulfite sequencing combines bisulfite conversion with targeted amplification of regions of interest, and the next-generation sequencing. It is a rapid and efficient tool for analysis of up to 10 kb of targeted genomic regions in up to 96 samples at a time. The results offer absolute quantitation of cytosine methylation at single-base resolution. Furthermore, this method can be used to confirm the results obtained from whole genome bisulfite sequencing, reduced representation bisulfite sequencing, and methylated DNA immunoprecipitation sequencing. Targeted bisulfite sequencing allows for better determination of SNPs and methylation status events, and a better understanding of human disease.
2. What are the advantages of targeted bisulfite sequencing as compared to other technologies of methylation sequencing?
Compared to whole genome methylation profiling studies, the methylation detection of only selected candidate regions is a more viable approach that makes methylation analysis more feasible and accessible in different settings. Targeted bisulfite sequencing dramatically increases data throughput and brings down the cost. Meanwhile, it is also able to detect the methylation status of regions that cannot be detected by RRBS (reduced representation bisulfite sequencing) or immunoprecipitation approaches. Targeted bisulfite sequencing has been widely applied to the methylation sequencing and validation of large sample cohort in target regions.
3. How to design proper probes or primers?
A successful application of targeted bisulfite sequencing largely depends on the proper design of probes and primers. There are a lot of effective tools for generating probes or primers in an automatic fashion, such as ppDesigner (http://genome-tech.ucsd.edu/public/Gen2_BSPP/ppDesigner/ppDesigner.php) and PRIMEGENSw3 (http://primegens.org). After obtaining many probes candidates or primers candidates, the validation process is important for selecting the most optimal ones.
4. What is the workflow of targeted bisulfite sequencing?
The workflow of PCR-based targeted bisulfite sequencing is depicted in Figure 1. As for hybridization-based targeted bisulfite sequencing, it can be performed either by bisulfite conversion of hybrid-selected native DNA or by hybrid selection of converted DNA. And the latter approach is now commercially available, which is characterized by superior target specificity, lower DNA-input requirements, and the ability to distinguish a C to U bisulfite conversion from a C to T SNP.
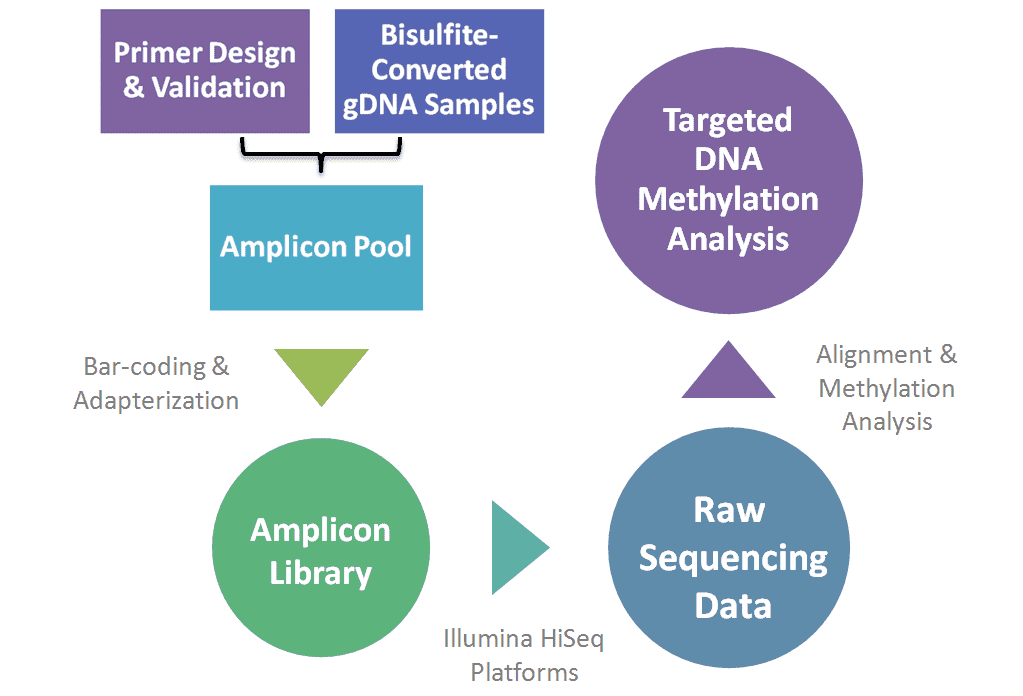 Figure 1. The workflow of PCR-based targeted bisulfite sequencing.
Figure 1. The workflow of PCR-based targeted bisulfite sequencing.
Targeted Bisulfite Seq Case Studies
Targeted methylation sequencing reveals dysregulated Wnt signaling in Parkinson disease
Journal: Journal of Genetics and Genomics
Impact factor: 4.051
Published: 2016 October 20
Background
Environmental and genetic factors play crucial roles in Parkinson disease etiology. For now, we have understood that a number of environmental toxins facilitate parkinsonism in humans and animals. Nevertheless, we have no idea about the potential interaction between genetics environment and genetics in Parkinson pathogenesis. Therefore, Zhang et al. apply both targeted bisulfite sequencing and immunohistochemistry methods to study the underlying links.
Methods
- Frozen tissues
- Formalin-fixed tissues
- Genomic DNA extraction
- Immunostaining
- Targeted DNA capture
- Shotgun library construction
- Bisulfite padlock sequencing
- Expression profiling array analysis
- Whole genome expression profiling
Results
1. Targeted bisulfite sequencing revealed increased methylation of genes in Wnt signaling
They designed a set of approximately 97,000 padlock probes covering 16,379 validated tissue differential methylation regions (T-DMRs), all known and predicted human imprinted genes and the transcription start sites of all genes on the X chromosome. K-mean clustering analysis yielded 336 non-overlapping DMRs, among which were mapped to known genes. And the majority of these regions (230/233) showed elevated levels of methylation in the PD brain tissues compared to healthy donors. There is a significant enrichment of genes involved in Wnt signaling and neurogenesis.
2. Reduced detection of genes in Wnt signaling in substantial nigra of PD patients
The levels of Foxc1, Ngn2, Spry1, and β-catenin in midbrain DA neurons were notably reduced in PD patients comparing to those of matched controls. Quantitative analysis revealed a significant increase of cells with reduced or no detectable expression of these genes. In contrast, the level of NEDD8 in the DA neurons of both PD patients and controls remain similar, suggesting that reduced detection of these proteins in midbrain DA neurons of PD patients is specific. Similar observations were made on Foxc1 and Spry1 in the cortical tissues of PD patients and their matched controls. However, little change of Ngn2 and β-catenin was detected in the cortical tissues of PD patients and their matched controls.
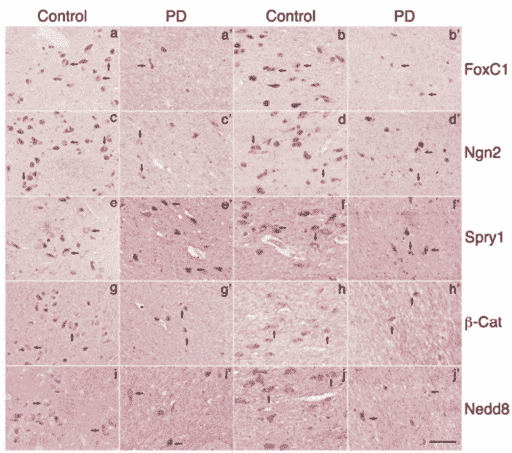 Figure 1. The immunodetection of Foxc1, Ngn2, Spry1, β-Catenin, and Nedd8 in human midbrain.
Figure 1. The immunodetection of Foxc1, Ngn2, Spry1, β-Catenin, and Nedd8 in human midbrain.
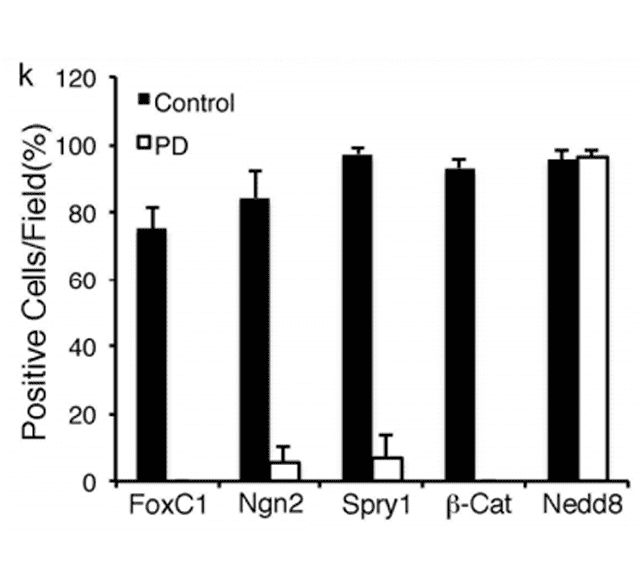 Figure2. Quantitative analysis revealed a significant increase of cells with reduced or no detectable expression of these genes.
Figure2. Quantitative analysis revealed a significant increase of cells with reduced or no detectable expression of these genes.
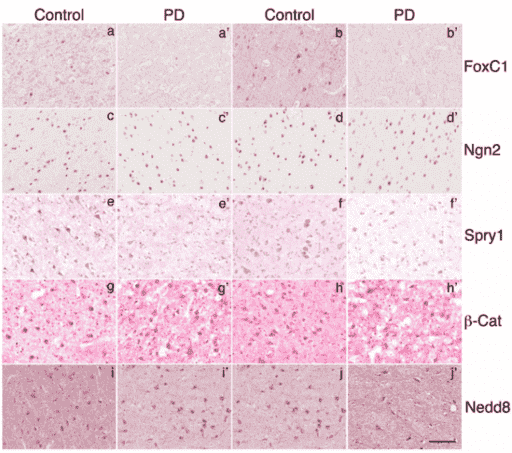 Figure 3. The immunodetection of Foxcl, Ngn2, Spry1, β-Catenin, and Nedd8 in human cerebral cortex.
Figure 3. The immunodetection of Foxcl, Ngn2, Spry1, β-Catenin, and Nedd8 in human cerebral cortex.
3. Down-regulation of Wnt signaling in cells treated with PD neurotoxin MPP+
Functional annotation analysis by MPP+ treatment indicates enrichment of up-regulated genes functioning in disulfide bond, cell membrane, neurological system process, defense response, cell adhesion, positive regulation of apoptosis and regulation of migration. MPP+-induced down-regulated annotation clusters include genes functioning in disulfide bond, secreted, extracellular matrix, plasma membrane, Wnt signaling and hedgehog signaling. The results further support a notion that Wnt signaling is impaired in neurotoxin-induced parkinsonism.
Reference:
- Zhang L, Jie D, Qian P, et al. Targeted methylation sequencing reveals dysregulated Wnt signaling in Parkinson disease. Journal of Genetics and Genomics, 2016, 43(10):587-592.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Restriction endonuclease cleavage of phage DNA enables resuscitation from Cas13-induced bacterial dormancy
Journal: Nature microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.


