
 Sample Submission Guidelines
Sample Submission Guidelines
RNA-Seq Variant Calling Pipeline: From Transcript Reads to Validated Mutations
RNA-Seq variant calling is a strong method for finding genetic changes in regions of the genome that are being actively transcribed. This paper outlines a clear workflow for finding germline and somatic mutations in RNA-sequencing data. It includes key steps like quality control, alignment, variant detection, filtering, and final validation. We examine cutting-edge computational tools and methodological best practices that address RNA-specific challenges including splicing junctions, RNA editing, and allele-specific expression. Additionally, we discuss strategies for distinguishing true genetic variants from technical artifacts and RNA processing events, thereby enhancing the accuracy and reliability of transcriptome-based variant identification for precision medicine applications.
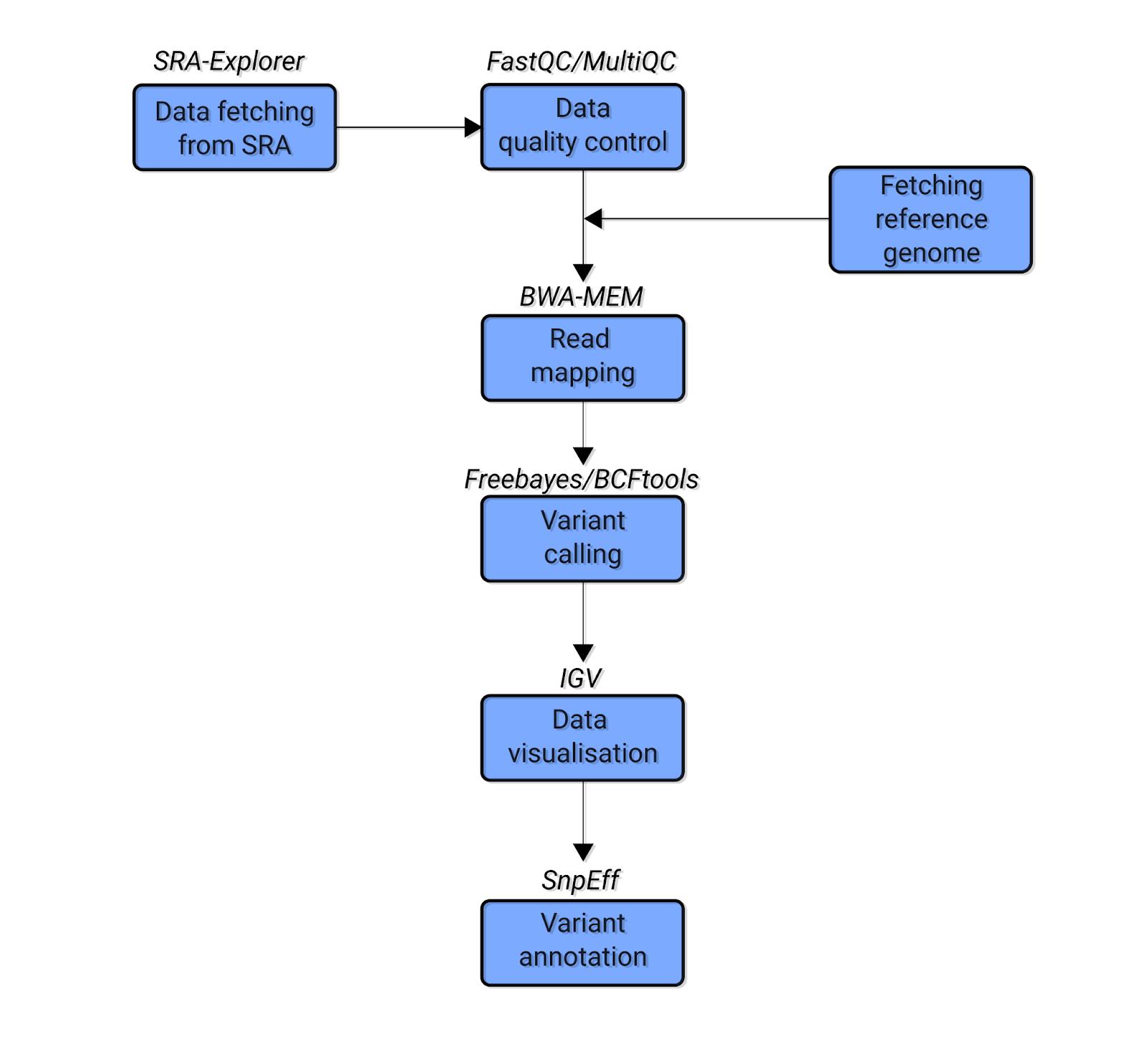 Figure 1. The simplified schematic bioinformatics pipeline of the variant calling analysis.Stepanka Zverinova, 2021)
Figure 1. The simplified schematic bioinformatics pipeline of the variant calling analysis.Stepanka Zverinova, 2021)
Data Quality Control and Preprocessing
Before embarking on variant calling, ensuring high-quality input data is crucial. RNA-Seq datasets often contain technical artifacts and biases that must be addressed through rigorous quality control and preprocessing steps.
Read Quality Assessment with FastQC
FastQC represents the gold standard tool for initial quality assessment of sequencing data. When applied to RNA-Seq reads, FastQC generates comprehensive reports that highlight potential issues including:
- Base Quality Scores: Phred quality scores typically decline toward the read ends. Scores below 20 (1% error rate) warrant attention.
- Sequence Content: Unusual patterns in nucleotide distribution may indicate adapter contamination or technical biases.
- GC Content: Deviations from expected GC distribution suggest potential contamination or library preparation biases.
- Sequence Duplication: Excessive duplication may indicate PCR artifacts or very high expression of specific transcripts.
- Overrepresented Sequences: Frequently occurring sequences often represent adapters, primers, or highly abundant transcripts like rRNA.
- K-mer Content: Unusual k-mer distributions can reveal biases in library preparation or sequencing.
A typical FastQC command for RNA-Seq analysis looks like:
bash
fastqc -o output_directory input_sample_R1.fastq.gz input_sample_R2.fastq.gz
Adapter and Quality Trimming with Trimmomatic
After quality assessment, preprocessing typically involves trimming low-quality bases and removing adapter sequences. Trimmomatic excels at these tasks, offering a range of options specific to RNA-Seq data:
bash
trimmomatic PE -phred33 input_R1.fastq.gz input_R2.fastq.gz \
output_R1_paired.fastq.gz output_R1_unpaired.fastq.gz \
output_R2_paired.fastq.gz output_R2_unpaired.fastq.gz \
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 \
SLIDINGWINDOW:4:15 MINLEN:36
The parameters in this command perform several key functions:
- ILLUMINACLIP: Removes adapter sequences specified in TruSeq3-PE.fa
- LEADING/TRAILING: Trims low-quality bases (below quality 3) from the start and end of reads
- SLIDINGWINDOW: Scans reads with a 4-base window, trimming when average quality drops below 15
- MINLEN: Discards reads shorter than 36 bases after trimming
RNA-Seq specific considerations during preprocessing include:
- rRNA Depletion: Ribosomal RNA can dominate RNA-Seq libraries. Tools like SortMeRNA can identify and remove rRNA reads.
- Poly-A Tails: Poly(A) tails in mRNA molecules can lead to low-complexity regions that should be trimmed.
- RNA Degradation: Degraded RNA samples may show 3' bias, requiring additional quality checks.
Technical Duplicate Handling
Unlike DNA-Seq, RNA-Seq data contains both PCR duplicates (technical artifacts) and natural duplicates (from highly expressed genes). For variant calling purposes, marking duplicates remains important, but complete removal could eliminate valuable signal from highly expressed genes. Tools like Picard MarkDuplicates can mark duplicates while preserving the information for downstream analysis:
bash
java -jar picard.jar MarkDuplicates \
I=input.bam \
O=marked_duplicates.bam \
M=marked_dup_metrics.txt
High-quality preprocessed data forms the foundation for accurate variant calling. The next step involves aligning these processed reads to the reference genome while accounting for the unique characteristics of RNA-Seq data.
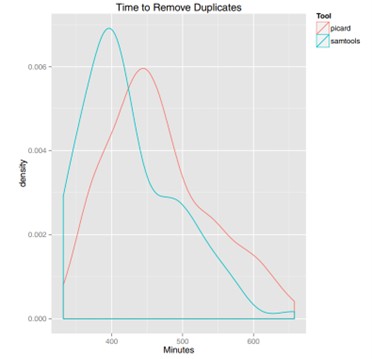 Figure 2. Density plot of execution time for both SAMTools and Picard duplicate removal.(Mark T W Ebbert, 2016)
Figure 2. Density plot of execution time for both SAMTools and Picard duplicate removal.(Mark T W Ebbert, 2016)
Services you may interested in
Learn More
Splice-Aware Alignment and Read Preparation
Splice-Aware Alignment
Accurate alignment of RNA-seq reads to a reference genome is a critical step in variant calling. Unlike DNA-seq reads, RNA-seq reads originate from mature mRNA transcripts where introns have been spliced out. Therefore, standard DNA aligners are not suitable for RNA-seq data as they cannot handle reads that span exon-exon junctions.
Splice-aware aligners such as STAR (Spliced Transcripts Alignment to a Reference) and HISAT2 (Hierarchical Indexing for Spliced Alignment of Transcripts) are specifically designed to handle the complexities of RNA-seq data. These aligners can map reads that span splice junctions by splitting them and aligning each segment to different exons.
STAR has become particularly popular due to its speed and accuracy. It builds a suffix array index of the reference genome and uses an algorithm that efficiently finds maximal mappable prefixes of each read. For a typical RNA-seq variant calling pipeline, STAR can be run with the following parameters:
bash
STAR--genomeDir /path/to/genome_index \
--readFilesIn sample_R1.fastq.gz sample_R2.fastq.gz \
--readFilesCommand zcat \
--outFileNamePrefix sample_ \
--outSAMtype BAM SortedByCoordinate \
--twopassMode Basic \
--outFilterMultimapNmax 20 \
--alignSJoverhangMin 8 \
--alignSJDBoverhangMin 1 \
--outFilterMismatchNmax 999 \
--outFilterMismatchNoverReadLmax 0.04 \
--alignIntronMin 20 \
--alignIntronMax 1000000 \
--alignMatesGapMax 1000000
The "two-pass" mapping approach is particularly beneficial for variant calling, as it first identifies splice junctions from the data and then uses this information to guide the final alignment.
Read Processing for Variant Calling
After alignment, several additional processing steps are required to prepare the data for variant calling:
- 1. Marking duplicates: Duplicate reads arising from PCR amplification can lead to false-positive variant calls. Tools like Picard's MarkDuplicates identify and mark these duplicates.
- 2. Splitting reads at N CIGAR operations: RNA-seq aligners represent reads spanning introns using the N operation in the CIGAR string. The GATK SplitNCigarReads tool splits these reads into multiple alignments, ensuring that only exonic segments are used for variant calling.
- 3. Base quality score recalibration (BQSR): This step adjusts the base quality scores to account for systematic errors in sequencing. For RNA-seq data, BQSR is performed using known variant sites as a training set.
Because RNA aligners have different conventions than DNA aligners, reformatting alignments that span introns is necessary for variant callers like HaplotypeCaller. The SplitNCigarReads step splits reads with N in the CIGAR into multiple supplementary alignments and hard clips mismatching overhangs. By default, it also reassigns mapping qualities for good alignments to match DNA conventions.
A typical GATK command for processing RNA-seq alignments might look like:
bash
gatk SplitNCigarReads \
-R reference.fasta \
-I input.bam \
-O split.bam
gatk BaseRecalibrator \
-R reference.fasta \
-I split.bam \
--known-sites known_variants.vcf \
-O recal_data.table
gatk ApplyBQSR \
-R reference.fasta \
-I split.bam \
--bqsr-recal-file recal_data.table \
-O recalibrated.bam
These preprocessing steps are essential for accurate variant calling, as they help to reduce artifacts and biases inherent to RNA-seq data. Proper alignment and processing of RNA-seq reads ensure that variants called in subsequent steps are genuine genetic differences rather than technical artifacts.
Variant Calling with RNA-Specific Parameters
Variant calling from RNA-seq data requires specialized approaches that account for the unique characteristics of transcriptomic data. Several tools have been developed or adapted for this purpose, with the Genome Analysis Toolkit (GATK) HaplotypeCaller and DeepVariant emerging as leading options.
GATK HaplotypeCaller for RNA-seq
GATK HaplotypeCaller uses a local de novo assembly approach to call variants, which is particularly beneficial for RNA-seq data where alignments around splice sites can be complex. The Genome Analysis Toolkit (GATK) developed at the Broad Institute provides state-of-the-art pipelines for germline and somatic variant discovery and genotyping.
When used for RNA-seq data, HaplotypeCaller requires specific parameters to optimize for the characteristics of transcriptomic data:
bash
gatk HaplotypeCaller \
-R reference.fasta \
-I recalibrated.bam \
-O variants.vcf \
--dont-use-soft-clipped-bases \
-stand-call-conf 20.0 \
--dbsnp dbSnp.vcf
The --dont-use-soft-clipped-bases parameter is particularly important for RNA-seq data, as it prevents the caller from using soft-clipped portions of reads, which might represent alignments across splice junctions rather than true variations.
DeepVariant for RNA-seq
Recently, deep learning-based approaches have shown promising results for variant calling from RNA-seq data. DeepVariant, a deep-learning-based variant caller, has been extended to learn and account for the unique challenges presented by RNA-seq data. The DeepVariant RNA-seq model produces highly accurate variant calls from RNA-sequencing data and outperforms existing approaches such as Platypus and GATK.
DeepVariant takes a fundamentally different approach to variant calling compared to traditional methods. It converts aligned reads into images and uses a convolutional neural network to identify variants, similar to image recognition. This approach allows the model to learn complex patterns in the data that might be difficult to capture with rule-based algorithms.
In benchmark comparisons, DeepVariant has shown superior performance for variant calling. In a comparison with GATK HaplotypeCaller, DeepVariant yielded a higher transition-to-transversion (Ti/Tv) ratio (2.38 ± 0.02) than GATK (2.04 ± 0.07), suggesting that DeepVariant proportionally called more true positives. The concordance rate between the two pipelines was 88.73%.
Somatic Variant Calling in RNA-seq
While the above methods are primarily designed for germline variant calling, RNA-seq data can also be used to identify somatic mutations, particularly in cancer research. For somatic variant calling, tools like MuTect2 (part of GATK) or Strelka2 can be adapted for RNA-seq data.
However, somatic variant calling from RNA-seq faces additional challenges, including:
- 1. Distinguishing somatic mutations from RNA editing events
- 2. Handling allele-specific expression that can skew variant allele frequencies
- 3. Accounting for tumor heterogeneity and the presence of multiple cell populations
Despite these challenges, RNA-seq can provide valuable insights into the expressed mutational landscape of tumors, complementing DNA-based approaches.
In recent years, there has been increased interest in developing joint calling approaches for RNA-seq data, similar to what is commonly used for DNA sequencing. The fully validated GATK pipeline for calling variants on RNA-seq data was traditionally a per-sample workflow that did not include joint genotyping analysis. Recent efforts have focused on combining modern GATK commands from distinct workflows to call variants on RNA-seq samples using joint genotyping.
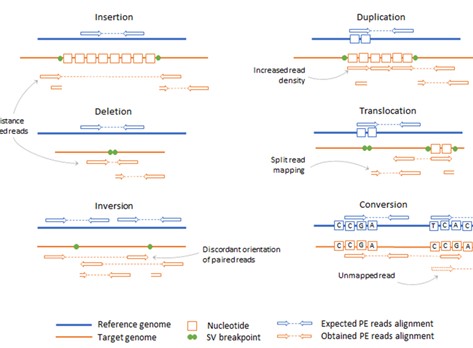 Figure 3. Diversity of DNA variant types.( Stepanka Zverinova, 2021)
Figure 3. Diversity of DNA variant types.( Stepanka Zverinova, 2021)
Filtering RNA-Specific Artifacts
Even with specialized variant calling methods, RNA-seq data can produce many false positive variant calls due to the inherent complexities of transcriptomic data. Therefore, rigorous filtering is essential to distinguish genuine variants from artifacts.
Common Sources of RNA-seq Artifacts
Several factors can lead to false positive variant calls in RNA-seq data:
- 1. RNA editing: Adenosine-to-inosine (A-to-I, read as A-to-G) and cytidine-to-uridine (C-to-U, read as C-to-T) editing are common post-transcriptional modifications that can be misinterpreted as genomic variants.
- 2. Alignment errors at splice junctions: Reads that span splice junctions may be incorrectly aligned, leading to false variant calls near the edges of exons.
- 3. Sequence-specific errors: Certain sequence contexts are prone to higher error rates during sequencing, resulting in systematic mismatches.
- 4. Reverse transcription errors: The conversion of RNA to cDNA during library preparation can introduce errors that appear as variants.
- 5. Low-complexity regions: Repetitive or low-complexity regions often have ambiguous alignments, leading to spurious variant calls.
Filtering Strategies
To effectively filter artifacts from RNA-seq variant calls, several strategies can be employed. These include excluding variants in RNA editing sites, filtering variants in low-complexity regions of coding sequences, and removing common genetic variants using databases like 1000 Genomes, gnomAD, and dbSNP.
A comprehensive filtering approach might include:
- 1. Filtering RNA editing sites: Known RNA editing sites can be obtained from databases like REDIportal and excluded from variant calls.
- 2. Expression-based filtering: Variants in genes with very low expression levels are more likely to be artifacts. Setting a minimum threshold for transcript expression (e.g., TPM > 1) can help filter out such variants.
- 3. Variant allele frequency (VAF) filtering: Due to allele-specific expression, genuine variants may not follow the expected 0.5 (heterozygous) or 1.0 (homozygous) allele frequencies seen in DNA sequencing. However, extremely low VAF values (e.g., < 0.2) in well-expressed genes may indicate artifacts.
- 4. Strand bias filtering: Genuine variants should be observed on both forward and reverse strands. A strong strand bias can indicate sequencing or alignment artifacts.
- 5. Distance from splice junction filtering: Variants called within a few bases of splice junctions are often artifacts. Excluding variants within 3-5 bases of known splice sites can improve accuracy.
Example GATK VariantFiltration command:
bash
gatk VariantFiltration \
-R reference.fasta \
-V variants.vcf \
-O filtered_variants.vcf \
--filter-name "StrandBias" \
--filter-expression "FS > 30.0" \
--filter-name "QualByDepth" \
--filter-expression "QD < 2.0" \
--filter-name "ReadPosRankSum" \
--filter-expression "ReadPosRankSum < -8.0" \
--filter-name "ClusteredEvents" \
--cluster-window-size 35 \
--cluster-size 3
In GATK's RNA-seq variant filtering, additional parameters are often included to filter out clustered events that may clutter result files, such as the cluster-size parameter which specifies the number of SNPs that make up a cluster (default value: 3).
Recent approaches have also leveraged machine learning to improve variant filtering in RNA-seq data. These methods can learn complex patterns that discriminate between true variants and artifacts, potentially improving upon rule-based filtering approaches.
 Figure 4. Analysis of Eip63E start-gained SNP in w1118; iso-2; iso-3.( Pablo Cingolani, 2012)
Figure 4. Analysis of Eip63E start-gained SNP in w1118; iso-2; iso-3.( Pablo Cingolani, 2012)
Validation and Benchmarking of RNA-Derived Variants
Validating variants called from RNA-seq data is crucial to ensure the reliability of findings. Several approaches are available for validation, ranging from orthogonal sequencing methods to benchmarking against reference datasets.
Cross-validation with Matched DNA-seq Data
The gold standard for validating RNA-seq variants is comparison with matched DNA sequencing data from the same individual. In clinical sequencing studies, variants are typically filtered to remove recurrent artifacts associated with short-read alignment and may be visually confirmed by manual review of the sequence alignments.
When comparing RNA-seq and DNA-seq variants, several patterns may emerge:
- 1. Concordant variants: Variants found in both RNA and DNA are likely genuine germline variants.
- 2. RNA-specific variants: Variants present in RNA but not in DNA may represent RNA editing events or somatic mutations in highly expressed genes.
- 3. DNA-specific variants: Variants present in DNA but not in RNA may occur in genes that are not expressed in the sequenced tissue or may be subject to allele-specific expression.
The concordance rate between RNA-seq and DNA-seq variants can vary depending on expression levels. Highly expressed genes typically show higher concordance, while lowly expressed genes may have more discrepancies due to insufficient coverage in RNA-seq data.
Benchmarking with Synthetic Datasets
While the above methods are primarily designed for germline variant calling, RNA-seq data can also be used to identify somatic mutations, particularly in cancer research. For somatic variant calling, tools like MuTect2 (part of GATK) or Strelka2 can be adapted for RNA-seq data.
However, somatic variant calling from RNA-seq faces additional challenges, including:
- 1. Distinguishing somatic mutations from RNA editing events
- 2. Handling allele-specific expression that can skew variant allele frequencies
- 3. Accounting for tumor heterogeneity and the presence of multiple cell populations
In the absence of matched DNA sequencing data, synthetic or reference datasets can be used for benchmarking variant calling pipelines. The Sequencing Quality Control 2 (SEQC2) project, led by the FDA, developed reference materials that could be shared by laboratories for standardized evaluation of NGS technologies. SEQC2 developed synthetic controls that provide an unambiguous representation of difficult sequences, including complex variants, which can be used to benchmark the performance of diverse sequencing technologies.
These synthetic datasets provide ground truth variant calls against which RNA-seq variant calling pipelines can be evaluated. Metrics commonly used for benchmarking include:
- 1. Sensitivity (Recall): The proportion of true variants that are correctly identified.
- 2. Precision: The proportion of called variants that are true variants.
- 3. F1 Score: The harmonic mean of precision and recall, providing a balanced measure of performance.
Benchmarking studies have shown that the DeepVariant RNA-seq model achieves the highest median F1 score across different genomic regions, performing the best overall in CDS regions. Additionally, runtime performance comparisons have shown that DeepVariant RNA-seq models are faster than GATK, while maintaining higher accuracy.
Integration with Functional Information
Another approach to validation is integrating variant calls with functional information. For example, variants that are predicted to have a significant functional impact (e.g., nonsense mutations, frameshift mutations) and occur in genes known to be associated with the phenotype under study are more likely to be genuine and biologically relevant.
Similarly, variants that show consistent patterns across multiple samples or are enriched in specific biological conditions may have higher confidence. For instance, recurrent mutations in cancer samples may represent driver mutations, even if they are not validated by DNA sequencing.
Visualization and Manual Review
Despite advances in automated filtering and validation methods, manual review of variant calls remains an important step in many studies, particularly for clinically relevant variants. Tools like the Integrative Genomics Viewer (IGV) allow researchers to visualize aligned reads supporting each variant, helping to identify potential artifacts or misalignments.
When reviewing RNA-seq variants in IGV, special attention should be paid to:
- 1. Read distribution: Variants should be supported by multiple independent reads.
- 2. Position within reads: Variants occurring predominantly at the ends of reads may be artifacts.
- 3. Splice junctions: Variants near splice junctions should be scrutinized carefully.
- 4. Base quality: Variants should be supported by high-quality base calls.
Visual confirmation through manual review of the sequence alignments remains an important validation step in clinical sequencing studies, where the accuracy of variant calls is paramount.
Conclusion
RNA-seq variant calling offers a valuable approach to identifying genetic variants within the transcribed portion of the genome. While it presents unique challenges compared to DNA sequencing, advances in bioinformatic methods and tools have significantly improved the accuracy and reliability of variant calling from RNA-seq data.
The pipeline described in this article—from quality control and preprocessing to alignment, variant calling, filtering, and validation—provides a comprehensive framework for extracting high-confidence variant calls from RNA-seq data. By leveraging RNA-specific parameters and filtering strategies, researchers can effectively distinguish genuine variants from technical artifacts.
Recent developments, particularly the application of deep learning approaches like DeepVariant, have pushed the boundaries of what is possible with RNA-seq variant calling. DeepVariant's RNA-seq model has demonstrated superior performance compared to traditional methods, highlighting the potential of machine learning to address the complex challenges inherent to transcriptomic data.
As sequencing technologies continue to evolve and computational methods improve, RNA-seq variant calling will likely become an increasingly valuable tool in genomics research, providing insights into the functional consequences of genetic variations and their role in human health and disease.
References:
- Zverinova, S., & Guryev, V. (2022). Variant calling: Considerations, practices, and developments. Human mutation, 43(8), 976–985. https://doi.org/10.1002/humu.24311
- Ebbert, M. T., Wadsworth, M. E., Staley, L. A., Hoyt, K. L., Pickett, B., Miller, J., Duce, J., Alzheimer's Disease Neuroimaging Initiative, Kauwe, J. S., & Ridge, P. G. (2016). Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC bioinformatics, 17 Suppl 7(Suppl 7), 239. https://doi.org/10.1186/s12859-016-1097-3
- Cingolani, P., Platts, A., Wang, leL., Coon, M., Nguyen, T., Wang, L., Land, S. J., Lu, X., & Ruden, D. M. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly, 6(2), 80–92. https://doi.org/10.4161/fly.19695


