
 Sample Submission Guidelines
Sample Submission Guidelines
RNA-Seq Variant Calling: Key Challenges and Emerging Solutions
RNA-Seqvariant calling is an effective way to find genetic changes in transcribed parts of the genome. Traditional DNA variant detection shows a broad view of genomic changes. RNA-Seq variant calling, however, highlights tissue-specific expression patterns and alternative splicing events. These can have important functional effects.
This paper looks at RNA-Seq variant calling. It helps find genetic changes in active genes. This method shows tissue-specific patterns and functional changes. It adds to the traditional DNA variant detection methods.
Introduction to RNA-Seq Variant Calling
RNA-Seq has mostly been used for expression profiling. But now, researchers see its value in finding genomic variants in expressed regions of the genome. RNA-Seq variant calling works well with traditional DNA methods for finding variants. RNA-Seq focuses on transcribed regions, unlike whole-genome or whole-exome sequencing. This approach has several key benefits for variant analysis. It offers better coverage of expressed genes. This could uncover important variants that DNA sequencing might miss at similar depths. RNA-Seq targets areas of the genome that are actively transcribed. This focus boosts the chances of finding variants that may have functional impacts. RNA-Seq lets researchers analyze genetic variation and gene expression at the same time. This helps them link genotype with transcriptional phenotypes directly.
RNA-Seq shows mutations in regions that are actively transcribed. This is different from DNA sequencing, which captures the entire genetic blueprint, no matter if the genes are expressed or not. This distinction offers several unique advantages:
- Mutations found in RNA-Seq are in expressed genes. So, they are more likely to have functional effects.
- Isoform-Specific Mutations: RNA-Seq shows mutations in specific transcript isoforms. This helps us understand changes in splice variants.
- Allele-Specific Expression: RNA-Seq can show allelic imbalance. This means one allele is expressed more than the other.
- Post-Transcriptional Modifications: RNA-Seq can reveal modifications that occur after transcription but before translation.
- Cost-Effectiveness: If RNA-Seq data is already made for expression analysis, you can do variant calling on that same dataset. This means no extra sequencing costs.
RNA-Seq Variant Calling's Applications
- Confirm the pathogenicity of variants of uncertain significance (VUS)
- Identify deep intronic variants that affect splicing
- Detect mutations in genes with tissue-specific expression patterns
- Reveal mutations in regulatory regions that affect expression levels
RNA-Seq variant calling is uniquely positioned to detect variants that affect splicing, including:
- Mutations in canonical splice sites
- Variants that create or destroy splicing enhancers or suppressors
- Alterations that lead to exon skipping or intron retention
- Mutations that activate cryptic splice sites
Despite these advantages, RNA-Seq variant calling presents unique challenges compared to DNA-based approaches. Identifying variants from RNA-Seq data is tough. This is due to intronic sequences, alternative splicing, RNA editing, and varying expression levels. A strong pipeline is key to overcoming these challenges and getting reliable variant information.
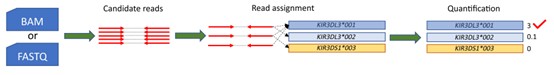 Figure 1. Overview of the T1K workflow.( Song, L, 2023)
Figure 1. Overview of the T1K workflow.( Song, L, 2023)
Services you may interested in
Learn More
RNA-Seq Variant Calling's Key Challenges
Low Coverage and Allelic Dropout in Low-Expressed Genes
RNA-Seq coverage is inherently variable and directly proportional to gene expression levels. Highly expressed genes can have thousands of reads. In contrast, lowly expressed genes usually have fewer reads. This sparse coverage makes it hard to detect variants in these areas. This uneven representation leads to several complications:
- Insufficient read depth to confidently call variants in low-expression regions
- Increased risk of false negatives due to inadequate coverage
- Allelic dropout, where one allele fails to be represented in the sequencing data
- Heterozygous variants can be wrongly classified as homozygous. This happens when there are no reads from one allele.
The challenge is clear in tissue samples with many cell types. Here, some variants may show up only in specific cell groups. Statistical methods can help with variable coverage and expression-based filtering. However, these issues are still big challenges in RNA-Seq variant calling.
Strand-Specific Biases and Reverse Transcription Artifacts
RNA-Seq library prep has several enzymatic steps. These steps can cause systematic biases and artifacts.
- Strand-specific protocols can create asymmetric coverage patterns between forward and reverse strands
- Reverse transcriptase enzymes can make mistakes while creating cDNA. This is especially true when they meet RNA secondary structures.
- Template switching during reverse transcription can generate chimeric cDNA molecules
- Sequence-specific pausing or premature termination of reverse transcription can create coverage gaps
- PCR amplification can cause errors. Some nucleotide contexts are more likely to have misincorporation.
These technical artifacts might be confused with real genetic variants. So, we need advanced filtering strategies. These strategies should look at strand bias, sequence context, and where the supporting reads are located.
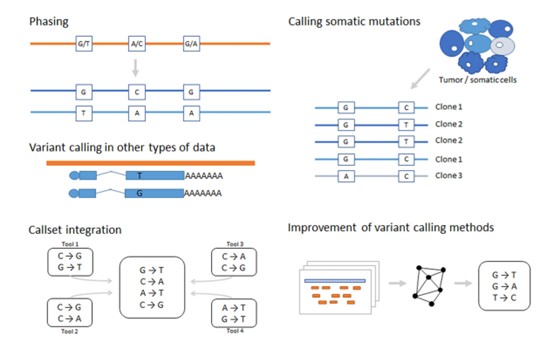
Figure 2. Current developments and challenges in variant identification technologies and algorithms. (Stepanka Zverinova, 2021)
Difficulty in Distinguishing True Mutations from RNA-Editing Events
RNA editing is a process that changes the RNA sequence after it is made. It does this without altering the DNA template. The most common type in humans is adenosine-to-inosine editing. This shows up as A-to-G changes in sequencing data and is done by ADAR enzymes. Other forms include cytidine-to-uridine editing (C-to-T) catalyzed by APOBEC enzymes.
These editing events pose significant challenges for RNA-Seq variant calling:
- RNA edits appear identical to genomic mutations in RNA-Seq data alone
- Editing can occur at thousands of sites throughout the transcriptome
- Editing efficiency varies across tissues, developmental stages, and physiological conditions
- Some sites get partially edited. This leads to a mix of edited and unedited transcripts.
Without matched DNA sequencing data, you can't easily tell true genomic variants apart from RNA editing events. This relies on:
- Characteristic sequence motifs surrounding known editing sites
- Databases of previously identified editing locations
- The ratio of variant to reference reads, which often differs between editing and genomic variants
- The type of nucleotide change, with A-to-G changes being more likely to represent editing than mutation
Advanced methods use these features and machine learning algorithms. These algorithms are trained on trusted editing sites. They help to better tell the difference between editing and mutation.
Emerging Solutions and Future Directions
Use of Single-Cell RNA-Seq to Detect Cell-Specific Expressed Variants
Single-cell RNA sequencing (scRNA-Seq)represents a paradigm shift in transcriptomics by enabling the analysis of gene expression and genetic variation at cellular resolution. This approach offers several advantages for variant calling:
- Detection of cell type-specific variants that might be diluted in bulk RNA-Seq
- Identification of somatic mutations present in subpopulations of cells
- Characterization of allelic expression patterns at single-cell resolution
- Linkage of geneticvariants to specific cellular phenotypes or states
Recent methodological advances have improved variant detection in scRNA-Seq data:
- Integration of information across cells with similar transcriptional profiles to boost detection power
- Computational approaches that account for technical dropouts and amplification biases
- Statistical frameworks specifically designed for the sparsity of single-cell data
Despite these advances, challenges remain, including limited coverage per cell, high dropout rates, and amplification biases. Ongoing developments in library preparation methods and computational tools continue to enhance the reliability of variant calling from single-cell data.
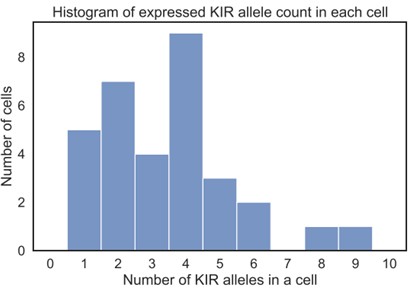
Figure 3. The number of expressed KIR alleles in a cell. (Song, L, 2023)
Long-Read Technologies for Resolving Complex Splicing
Traditional short-read RNA-Seq technologies are limited in their ability to resolve complex splicing patterns and detect variants within alternatively spliced regions. Long-read sequencing platforms, such as Pacific Biosciences (PacBio) Iso-Seq and Oxford Nanopore Technologies (ONT), overcome these limitations by generating reads that span entire transcripts:
- Full-length transcript sequencing eliminates ambiguities associated with splice junction mapping
- Direct observation of exon connectivity improves detection of variants in alternatively spliced regions
- Long reads enable phasing of multiple variants within the same transcript
- Better resolution of repetitive regions that are challenging for short-read technologies
These advantages are particularly valuable for:
- Detecting variants that affect splicing patterns
- Identifying fusion transcripts and complex structural variants
- Characterizing isoform-specific variants
While long-read technologies have historically been limited by higher error rates, recent improvements in sequencing chemistry and base-calling algorithms have substantially increased accuracy. Hybrid approaches that combine the high accuracy of short reads with the structural insights from long reads represent a promising direction for comprehensive variant calling.
Graph-Based Aligners and Machine Learning Tools for Low-Frequency Variant Detection
Traditional variant calling approaches rely on linear reference genomes and position-based alignments, which are suboptimal for capturing the full spectrum of human genetic diversity. Two emerging technologies are transforming this landscape:
Graph-based aligners replace linear references with graph structures that incorporate known genetic variations:
- Improved alignment accuracy near structural variants and complex genomic regions
- Reduced reference bias for populations divergent from the standard reference
- Better handling of insertions, deletions, and complex structural variants
- Enhanced ability to represent and detect population-specific variants
Machine learning and deep learning approaches leverage multiple features to distinguish true variants from technical artifacts:
- Integration of sequence context, base quality, mapping quality, and other features for variant classification
- Ability to recognize subtle patterns associated with true variants versus sequencing errors
- Adaptation to dataset-specific characteristics through training
- Enhanced sensitivity for detecting low-frequency variants
Tools like DeepVariant, which employ convolutional neural networks to analyze "images" of aligned reads, have demonstrated superior performance for DNA variant calling and are being adapted for RNA-Seq applications. These computational advances, combined with increasing data volumes for training, promise to substantially improve the detection of low-frequency variants from RNA-Seq data.
The convergence of these emerging technologies—single-cell resolution, long-read sequencing, graph-based alignment, and machine learning—heralds a new era in RNA-Seq variant calling, enabling more comprehensive, accurate, and functionally relevant characterization of genetic variation in expressed genes.
Conclusion
Variant calling from RNA-Seq data is a strong but tough way to find genomic changes in active parts of the genome. RNA-Seq data has unique challenges. These include variable coverage, allelic dropout, strand-specific biases, and RNA editing. Because of this, we need special methods. Regular DNA-based variant calling won't work here. RNA-Seq variant calling has clear benefits. It targets active regions and captures the unique genetic complexity of transcripts.
The field is rapidly evolving, driven by technological and computational innovations. Single-cell RNA-Seq technologies are revealing new layers of cell diversity. Also, long-read sequencing platforms are providing clear insights into complex transcriptome structures. Computational advances in graph-based alignment and machine learning are boosting variant detection. They improve both sensitivity and specificity. This is especially true for low-frequency variants that traditional methods might miss.
As these technologies develop and link up, we can look forward to a deeper understanding of how genetic variation impacts phenotypic expression. The future of RNA-Seq variant calling is more than spotting mutations. It's about placing these mutations in the larger context of gene expression, splicing dynamics, and cellular diversity. This integrated perspective will be instrumental in advancing our understanding of human genetics, disease mechanisms, and personalized medicine approaches.
For researchers and clinicians alike, staying abreast of these developments is essential. The choice of appropriate methodologies and analytical pipelines should be guided by the specific research questions, sample characteristics, and available resources. As the field continues to evolve, the integration of multiple approaches—combining the strengths of different sequencing technologies, computational methods, and validation strategies—will likely yield the most comprehensive and reliable insights into the complex world of expressed genetic variations.
Reference:
- Song, L., Bai, G., Liu, X. S., Li, B., & Li, H. (2023). Efficient and accurate KIR and HLA genotyping with massively parallel sequencing data. Genome research, 33(6), 923–931. https://doi.org/10.1101/gr.277585.122
- Zverinova, S., & Guryev, V. (2022). Variant calling: Considerations, practices, and developments. Human mutation, 43(8), 976–985. https://doi.org/10.1002/humu.24311


