
 Sample Submission Guidelines
Sample Submission Guidelines
Quality Control of ATAC Sequencing Library
ATAC-seq (Assay for Targeting Accessible-Chromatin with high throughout sequencing) is a powerful technique that leverages the transposition properties of the Tn5 enzyme to investigate open chromatin, transcription factor imprinting, and nucleosome localization. However, due to the considerable time and cost involved in sequencing, researchers and sequencing companies often conduct pre-sequencing quality checks on the constructed libraries. These checks encompass various parameters, such as library concentration and fragment size, to ensure reliable and meaningful results.
Recommended Reading: ATAC-Seq – A Method to Study Open Chromatin.
Compared to other sequencing libraries, ATAC-seq libraries pose unique challenges in terms of quality assessment. The uncertainty arises from potential variations between sample types and pre-treatments, which can significantly impact the quality of the generated libraries.
ATAC-seq libraries are typically prepared from intact nuclei or cells, where the transposon penetrates the nuclear or cellular membrane to access the chromatin's interior. The transposition reaction then incorporates sequencing junction sequences into the fragmented DNA. However, the accessibility of chromatin can be influenced by several factors, including cell type, cell number, cell activity, cell clustering, and the relative concentration of the transposon to DNA. As a result, the number and size of library fragments may exhibit considerable variability.
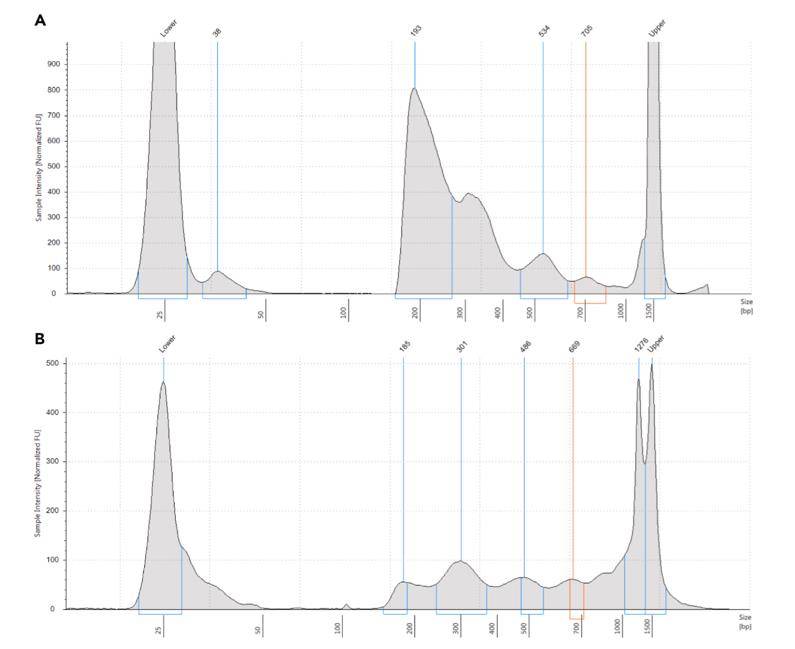 Typical TapeStation profile (D1000 TapeStation in this case) of an ATAC-seq library. (Maor-Nof et al., 2021)
Typical TapeStation profile (D1000 TapeStation in this case) of an ATAC-seq library. (Maor-Nof et al., 2021)
To address these challenges and ensure reliable data, comprehensive quality control measures are indispensable. Researchers meticulously assess the library concentration and fragment size distribution to optimize sequencing efforts. Moreover, understanding the specific characteristics of each sample and accounting for potential variations can aid in interpreting the resulting data accurately.
Fragment Size Analysis for ATAC-Seq Libraries
For a comprehensive evaluation of ATAC-Seq libraries, it is essential to analyze the distribution of DNA fragments post-library amplification. We highly recommend employing a DNA fragment analyzer to achieve this task. By generating an electropherogram of the fragments, researchers can gain valuable insights into the fragment sizes and their peak shapes. To ensure accurate results, it is advisable to utilize an assay with a resolution of less than 1000 base pairs (bp).
The fragment size analysis provides critical information about the library's integrity and efficiency. A well-defined and concentrated peak in the electropherogram signifies a homogeneous distribution of DNA fragments within the desired range. Conversely, irregular peak shapes or broad size distributions may indicate potential issues with library preparation or amplification.
By conducting this analysis, researchers can confidently assess the success of the ATAC-Seq library preparation and make informed decisions regarding the sequencing strategy. Ultimately, this step contributes significantly to the reliability and quality of the generated data, leading to more accurate interpretations of chromatin accessibility and regulatory element mapping.
Quality Control of ATAC-Seq Libraries
The fundamental unit of eukaryotic chromatin is the nucleosome, comprising 147 base pairs (bp) of DNA wrapped around a histone octamer, connected by 20-90 bp DNA linkers. The compactness of chromatin, determined by its interaction with histones, plays a crucial role in regulating gene expression.
In regions of open chromatin, where DNA weakly interacts with histones, transcription promoters and enhancers can easily bind to DNA, facilitating transcription. Conversely, in closed chromatin regions with tight DNA-histone binding, transcription factors struggle to access promoter and enhancer regions, inhibiting transcription.
ATAC-Seq capitalizes on the modified Tn5 enzyme's ability to access open chromatin regions, cleaving DNA into fragments while adding the required adapter sequences for sequencing. The transposition process can occur at various junction regions between nucleosomes, resulting in short fragments (<90 bp) or larger fragments with nucleosomes sandwiched between Tn5-cut DNA.
Following library amplification and addition of Illumina indexes, the library fragments typically range from approximately 200 bp to 1000 bp. Peaks between 160-200 bp appear due to the periodicity of neighboring nucleosomes.
The size and shape of ATAC-Seq library fragments can vary significantly between samples due to differences in cell types, activity status, cell numbers, and sample processing. This variability depends on cell-specific characteristics and the ratio of transposons to DNA.
The variation in nucleosome peaks is influenced by the frequency of DNA cleavage by Tn5, but the number or size of peaks does not always correlate directly with sequencing data quality. Library peak shapes often resemble a ski slope rather than displaying distinct peaks.
However, the most critical factor for libraries is to exhibit a good distribution of fragments in the 200-1000 bp range, preferably below 600 bp. This characteristic ensures the capture of essential chromatin accessibility information and enhances the reliability of the ATAC-Seq data.
Optimizing Transposition Reaction for Efficient ATAC-Seq Library Preparation
The transposition reaction is a critical step in ATAC-Seq library preparation that involves the addition of the Tn5 enzyme to access chromatin and cleave DNA into fragments. However, insufficient Tn5 or low Tn5-to-DNA ratio can lead to inadequate transposition, resulting in under-tagmentation. This phenomenon is characterized by major DNA fragments concentrated around 800bp.
Large fragments pose several drawbacks, such as inefficient clustering on flow cells, leading to lower clustering density and reduced sequencing output. To achieve the desired coverage, more sequencing is required for larger fragments compared to smaller ones.
Under-tagmentation can stem from various factors, with inadequate nucleus preparation being a common cause. Obtaining well-separated and suspended nuclei from tissue samples is more challenging than from cultured cells, making under-tagmentation more prevalent in tissue samples.
To address under-tagmentation issues, the following suggestions can be helpful:
- Ensure complete resuspension in lysis buffer and homogenize tissue samples for optimal nucleus preparation.
- Extend cell lysis time on ice to enhance Tn5 activity.
- Increase transposase reaction time to promote efficient cleavage.
- Add an extra PBS wash before lysis to eliminate potential inhibitors of lysis in the sample.
Another cause of oversized DNA fragments could be poor sample quality, particularly when dead cells release excessive amounts of open DNA in the cell suspension. This excess DNA can saturate Tn5 activity, reducing its binding to chromatin DNA.
To avoid this issue, it is recommended to use fresh cells with high activity or cells frozen in a solution of 50% FBS, 40% growth medium, and 10% DMSO. For tissue samples, snap-freezing in liquid nitrogen and storing at -80℃ is essential to preserve sample quality. Experiments should be conducted swiftly, and tissue should not be allowed to thaw before mincing in a petri dish on ice.
In cases where FACS-sorted cells are used, caution should be exercised to minimize cell damage during the sorting process. Employ sorting techniques optimized to reduce damage and aim to start with as many cells as possible. Additionally, sorting rare cell populations with less than 10% occupancy can cause more damage than sorting cells with high occupancy (e.g., 70%).
Apart from the mentioned precautions, precise experimental manipulation and attention to detail during the experimental process are essential for minimizing inter-sample errors.
Library Yields of ATAC-Seq
When starting with 50,000-100,000 fresh cells, an ideal output of 400-600 ng can be achieved. This output is obtained in a final elution volume of 20 μL, with a concentration of approximately 20-30 ng/μL.
However, it is important to note that FACS-sorted cells may yield lower amounts of DNA due to cell damage during the sorting process. Additionally, if it's the first time attempting the ATAC-Seq experiment, the unfamiliarity with the steps may lead to DNA loss during experimental operations.
To minimize sample loss, especially when working with SPRI magnetic beads, a gentle approach is key. Ethanol should be gently added to the bottom of the tube, allowing it to rise slowly without disturbing the magnetic beads. Over-drying the beads should be avoided, as it can cause bead breakage and significant sample loss. During DNA column purification, adding the elution buffer to the center of the column ensures efficient DNA recovery.
For ATAC-Seq experiments, relatively small amounts of DNA are required for sequencing. Therefore, while yield is important, it is not necessary to overly focus on it. As long as there is sufficient DNA for sequencing and the library fragments appear satisfactory, the libraries can proceed to be sequenced.
Variability in ATAC-Seq Library
It is common to observe differences in peak shapes among ATAC-Seq library fragments from various experimental samples and even between different batches of the library preparation process. These variations can arise due to factors such as the number of cells used, cell lysis conditions, or other subtle differences that influence the cleavage frequency of Tn5 or pA-Tn5.
Despite such variations in cleavage frequency and resultant differences in the appearance of library fragments, the sequencing data quality can remain stable and reliable across these samples. This stability is primarily due to the fact that the data peaks are largely governed by the accessibility of Tn5 to open chromatin or pA-Tn5 to antibodies.
While the appearance of the library's peak shapes can serve as a qualitative indicator to decide whether to proceed with sequencing, it is essential to understand that the quality of the library alone does not guarantee sequencing success. However, the results of library quality control (QC) can provide valuable insights into the likelihood of sequencing success and offer opportunities for process optimization based on QC outcomes.
To ensure accurate quantification of the library, qPCR-based assays should be utilized. This step further enhances the confidence in the library's suitability for sequencing and facilitates informed decisions in the sequencing workflow.
In conclusion, understanding the variability in ATAC-Seq library fragment peaks is crucial for interpreting sequencing results effectively. While peak appearance can guide the decision-making process, comprehensive library QC, along with qPCR-based quantification, contributes to the successful execution of ATAC-Seq experiments and the generation of meaningful epigenetic insights.
Reference:
-
Beck, Daniel, Millissia Ben Maamar, and Michael K. Skinner. "Genome-wide CpG density and DNA methylation analysis method (MeDIP, RRBS, and WGBS) comparisons." Epigenetics 17.5 (2022): 518-530.


