
 Sample Submission Guidelines
Sample Submission Guidelines
Applications of Pan Genome in Plants
Pan-genome analysis represents a paradigm shift in plant genomics, enabling comprehensive characterization of species-level genetic diversity. By combining core and variable genomic regions, it overcomes limitations of single-reference genomes. We discuss pan-genome methodology and its applications in studying plant genetic variation, domestication history, breeding improvement, and functional genomics.
What is Pan-Genome?
The pan-genome represents the complete genomic repertoire of a species, comprising both conserved core sequences (universally present across all accessions) and variable accessory components (present in specific lineages). Originally developed for prokaryotic genomics, this framework has been successfully adapted to eukaryotic systems, with pioneering plant pan-genome studies emerging from soybean research. Subsequent investigations have expanded pan-genomic analyses to major crops including Oryza sativa, Solanum lycopersicum, and Triticum aestivum. This approach proves particularly valuable for plant systems given their extensive structural variations, frequent polyploidization events, and abundant repetitive elements - features that conventional single-reference genomes often fail to adequately represent.
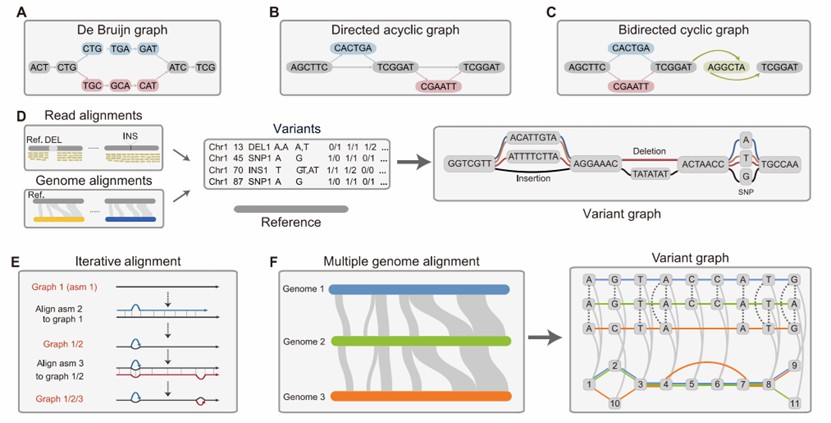 Figure 1. Pangenome models and construction of a pangenome graph.(Ze-Zhen Du, et. al,2025)
Figure 1. Pangenome models and construction of a pangenome graph.(Ze-Zhen Du, et. al,2025)
Services you may interested in
Learn More
Principles of Pan-Genome
Construction of Pan-Genome
Pan-genome construction requires the systematic integration of multiple genome assemblies through two principal methodologies: reference-guided and de novo strategies. The reference-based method aligns constituent genomes against an established reference sequence, subsequently incorporating detected variants into the pan-genome structure. This approach demonstrates optimal utility when leveraging high-quality reference genomes as organizational scaffolds. Alternatively, the de novo methodology independently assembles individual genomes prior to their consolidation into a comprehensive pan-genome. While computationally intensive, this strategy proves particularly effective for genetically diverse taxa or when reference genomes are unavailable. Selection between these approaches necessitates careful consideration of existing genomic resources and available computational capacity.
2.2 Graph-Based Representation
Graph-based pan-genomes represent genetic variations as nodes and edges in a graph structure. This approach allows for the efficient integration of multiple genomes and the representation of complex variations, such as structural variants (SVs) and presence-absence variations (PAVs). Tools like Minigraph and PGGB have been developed to construct graph-based pan-genomes, enabling more accurate read mapping and variant calling compared to traditional linear pan-genomes. Graph-based pan-genomes can handle multiple haplotypes and complex genomic regions more effectively, making them particularly useful for plant genomes with high levels of repetitive sequences and structural variations.
2.3 Variant Calling and Genotyping
Pan-genomes facilitate the detection of a wide range of genetic variants, including single nucleotide polymorphisms (SNPs), insertions/deletions (indels), and SVs. Variant calling in pan-genomes involves aligning sequencing reads to the pan-genome graph and identifying differences between the reads and the reference sequences. Genotyping assigns specific alleles to individuals based on the detected variants. Tools like vg, GraphTyper, and BayesTyper have been developed to perform variant calling and genotyping in pan-genome contexts. These tools can handle complex variations and provide more accurate genotyping results, especially in regions with high genetic diversity and repetitive sequences.
Applications of Pan-Genome in Plants
Case Study: Pan-Genome Analysis in Rice
In a groundbreaking study published in Genome Biology, Wang et al. (2023) developed a novel pangenome analysis pipeline called PSVCP (Presence/Absence Variation Calling Pipeline) to construct a high-quality rice pangenome and explore its applications in functional gene identification. This case study highlights the potential of pan-genome analysis in uncovering genetic diversity and improving crop breeding.
Background
Rice (Oryza sativa L.) is a staple food for nearly half of the world's population. To meet the increasing demand for food production amidst climate change, it is crucial to enhance rice productivity through genomics-assisted breeding. Traditional genetic analysis using a single reference genome often leads to biases, especially when dealing with significant structural variations (SVs) between individuals. Pan-genomes, which represent the complete genetic diversity within a species, offer a more comprehensive approach to capturing genomic variations.
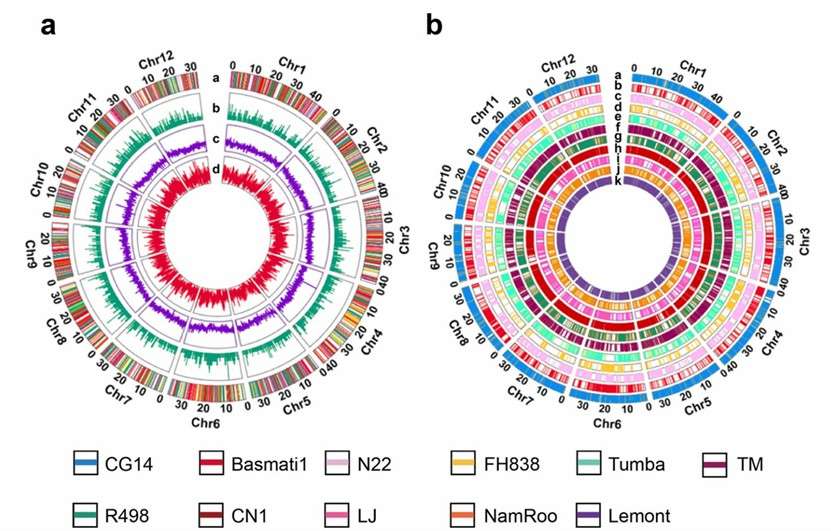 Figure 2. Feature of the rice pangenome.(Jian Wang, et. al,2023)
Figure 2. Feature of the rice pangenome.(Jian Wang, et. al,2023)
Methods
The researchers used an iterative alignment strategy to construct a linearized rice pangenome with 12 representative rice genomes. They then mapped short-read sequencing data from 413 diverse rice accessions to this pangenome to detect presence/absence variations (PAVs), translocations, and inversions. The pipeline involved three main steps: (1) identifying novel segments and integrating them into the reference genome, (2) mapping sequencing reads to the pangenome to detect PAVs, and (3) calling SVs based on the pangenome.
Results
The constructed rice pangenome included 24,585 novel sequences, with 1250 potential translocations and 3326 inversions. The average mapping rate to the pangenome was 97.84%, significantly higher than the 93.05% mapping rate to the Nipponbare reference genome. This indicated that the pangenome captured more genetic diversity than a single reference genome. The study identified 11,617 dispensable genes across the 413 rice accessions, with functions related to photosynthesis, defense response, and pathogenesis.
Functional Gene Identification
The researchers conducted a genome-wide association study (GWAS) using both SNPs and PAVs. For thousand grain weight (TGW), the SNP-GWAS identified 354 significant associations, but the PAV-GWAS pinpointed the causal variations directly. Similarly, for plant height (PH), the PAV-GWAS identified a novel locus (qPH8-1) not detected by SNP-GWAS. This locus contained a 13-kb insertion associated with significant differences in plant height.
Conclusions
The PSVCP pipeline demonstrated the power of pan-genome analysis in capturing genetic diversity and identifying functional genes. The rice pangenome constructed in this study provides valuable resources for future rice genomics research and breeding. The ability to identify causal PAVs for important agronomic traits highlights the potential of pan-genome approaches in accelerating crop improvement and addressing global food security challenges.
Case Study: Evolution and Functional Analysis of the Annexin Gene Family in the Maize Pan-Genome
Pan-genomes have been instrumental in identifying functional loci associated with crop domestication and breeding. For instance, a 10 kb PAV in a soybean genome was found to control seed cluster variation. In rice, SV-based genome-wide association studies (GWAS) have identified candidate functional SVs linked to traits like leaf senescence. These studies demonstrate that pan-genomes can capture genetic variations missed by conventional approaches, thereby enhancing breeding efforts and understanding genetic diversity within plant species. By incorporating pan-genome data into breeding programs, researchers can identify and select desirable traits more effectively, leading to improved crop varieties with enhanced yield, disease resistance, and stress tolerance.
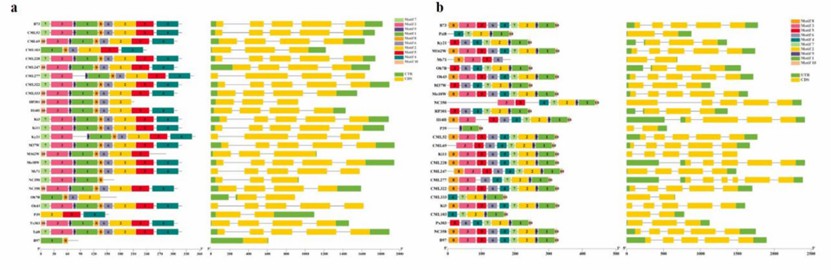 Figure 3. Gene structure of ZmAnn3 (a) and ZmAnn10 (b) in 26 maize genomes. (Liu, X, et. al,2025)
Figure 3. Gene structure of ZmAnn3 (a) and ZmAnn10 (b) in 26 maize genomes. (Liu, X, et. al,2025)
Background
Annexins (Anns) are a family of calcium-dependent, phospholipid-binding proteins that play critical roles in plant growth, development, and stress responses. Understanding the genetic mechanisms underlying these functions is essential for improving crop productivity and stress tolerance. This study leverages the pan-genome of 26 high-quality maize genomes to comprehensively analyze the annexin gene family, including their evolutionary patterns, expression profiles, and roles in stress responses.
Methods
The researchers used HMMER and Blastp to identify annexin genes in the maize pan-genome, identifying 12 ZmAnn genes, including 9 core genes and 3 near-core genes. Phylogenetic analysis was performed using a neighbor-joining model, and the Ka/Ks values were calculated to assess selective pressures. The study also analyzed cis-regulatory elements and structural variations (SVs) in the promoter regions of these genes. Transcriptome data from various maize tissues under cold stress were used for expression analysis and co-expression network construction.
Results
The study identified 12 ZmAnn genes, with 9 core genes in all 26 maize lines and 3 near-core genes in 24–25 lines. The Ka/Ks analysis revealed that ZmAnn10 was under positive selection in certain varieties, while the remaining genes showed purifying selection. Phylogenetic analysis divided ZmAnn proteins into six groups, with group VI containing only ZmAnn12. Structural variations were found to alter conserved domains, generating atypical genes. Transcriptome analysis showed distinct expression patterns for different Ann members in various tissues and under different stress treatments. Weighted gene co-expression network analysis identified four Ann genes (ZmAnn2, ZmAnn6, ZmAnn7, ZmAnn9) involved in co-expression modules under cold stress.
Conclusions
This study comprehensively analyzes the annexin gene family in maize, highlighting their evolutionary conservation and functional diversity. Identifying core and near-core genes and insights into their expression patterns and roles in stress responses underscores the importance of pan-genome analysis in uncovering genetic diversity and functional mechanisms. The findings suggest that certain ZmAnn genes, such as ZmAnn2 and ZmAnn7, play significant roles in cold stress tolerance, potentially offering new targets for breeding maize varieties with enhanced stress resistance.
Challenges and Future Directions
Computational Complexity
Constructing and analyzing pan-genomes require significant computational resources, especially for large and complex plant genomes. Developing more efficient algorithms and tools tailored for plant genomes is essential to address this challenge. Current tools often struggle with the high memory usage and long run times required for constructing pan-genome graphs and performing variant calling and genotyping. Future advancements in computational methods and hardware will be crucial for scaling up pan-genome analyses to include thousands of genomes.
Handling Structural Variations
Accurate detection and representation of SVs in pan-genomes remain challenging due to their complexity and the presence of repetitive sequences. Improving the resolution and accuracy of SV detection will enhance pan-genomes' utility in plant genomics. Current methods often fail to fully capture SVs in repetitive regions, leading to incomplete or inaccurate representations of genetic diversity. Developing new bioinformatics tools and techniques that can better handle repetitive sequences and complex SVs will be essential for advancing pan-genome studies in plants.
Integration with Other Omics Data
Integrating pan-genome data with other omics data, such as transcriptomics and epigenomics, will provide a more comprehensive understanding of plant biology. This integrative approach will help elucidate the functional impact of genetic variations and their roles in plant development and stress responses. Current efforts in multi-omics integration are still in their infancy, and more work is needed to develop robust methods for combining and analyzing these diverse datasets. Future studies should focus on developing integrative bioinformatics pipelines that can leverage the full potential of pan-genome data in combination with other omics data.
Conclusion
Pan-genomes have emerged as a powerful tool in plant genomics, offering a more comprehensive representation of genetic diversity compared to traditional single-reference genomes. They have facilitated the discovery of previously hidden genetic variations, provided insights into crop domestication and breeding, and enabled the identification of functional genes. Despite the challenges, ongoing advancements in sequencing technologies and bioinformatics tools will continue to enhance pan-genomes' applications in plant research and breeding, ultimately contributing to global food security and sustainable agriculture. Future work should focus on developing more efficient computational methods, improving the accuracy of SV detection, and integrating pan-genome data with other omics data to gain a holistic understanding of plant biology.
References:
- Du, ZZ., He, JB. & Jiao, WB. Plant graph-based pangenomics: techniques, applications, and challenges. aBIOTECH (2025). https://doi.org/10.1007/s42994-025-00206-7
- Liang, W., Zhao, Y., et.al., (2019). Non-invasive diagnosis of early-stage lung cancer using high-throughput targeted DNA methylation sequencing of circulating tumor DNA (ctDNA). Theranostics, 9(7), 2056–2070. https://doi.org/10.7150/thno.28119
- Liu, X., Zhang, M., et, al. (2025). The evolution, variation, and expression patterns of the annexin gene family in the maize pan-genome. Scientific reports, 15(1), 5711. https://doi.org/10.1038/s41598-025-89119-5


