
 Sample Submission Guidelines
Sample Submission Guidelines
A Review of Crop Pangenome
With the continuous development of genome sequencing technology, more and more species have completed the deciphering of the whole genome code, which is important for in-depth research on the functional gene localization and domestication of a species based on the comprehensive analysis of genome information. However, during the long evolutionary process of species, due to the influence of natural and human selection, each individual has developed extremely unique genetic traits, and the reference genome of a single individual can no longer cover all the genetic information of the species. In other words, if only a single reference genome is used for the study of genetic variation, a lot of intentional genetic code information may be lost, because many unique sequences are not on the reference genome, and because the price of gene sequencing has become cheaper, it has provided the possibility of pan-genome studies, especially in crop studies such as rice, maize, soybean, tomato, cotton, rape, Arabidopsis, etc. It has gradually become universal.
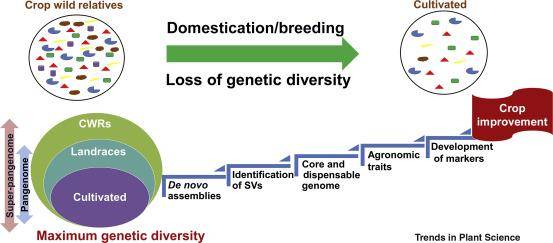 The concept of the pangenome and super-pangenome and their use for crop improvement. (Khan et al., 2020)
The concept of the pangenome and super-pangenome and their use for crop improvement. (Khan et al., 2020)
History of Pan-genome
Pan-genome is a general term for all genes of a species, which is distinct from the genes of individual genomes. In 2005, Tettelin H et al. first proposed the concept of microbial pangenome (pan from the Greek 'παν' meaning all), which is a general term for all genes of a species. In 2009, Li et al. first used the new whole-genome assembly method to splice multiple human genomes and discovered the unique DNA sequences and functional genes of individuals, and first proposed the concept of "human pan-genome", i.e., the sum of genetic sequences of human populations. In 2013, pangenome sequencing was applied to plant and animal research; in 2014, pangenome research of crops, such as soybean, rice, maize, oilseed rape, cotton, etc., was started.
The Main Research Content of Pan-Genome Studies
The main research content of pan-genome studies involves the analysis and characterization of the core genome and dispensable genome of plant and animal strains. The core genome consists of genes that are present in all strains, and they typically control basic metabolic functions in organisms. On the other hand, the dispensable or variable genome includes genes that are present in one or more strains, and they can contribute to diverse traits such as disease resistance or cold resistance.
Pan-genome research focuses on understanding the structural variation within the dispensable genome. Structural variations refer to differences in the arrangement, size, or presence/absence of genetic material, such as duplications, deletions, inversions, or insertions. These structural variations can have significant implications for the phenotypic diversity observed in individuals.
To study structural variation within the pan-genome, researchers often employ long read sequencing technologies such as PacBio SMRT or Nanopore Technology. These technologies offer advantages in terms of genome assembly and the detection of structural variations. They can provide long reads, which enable the assembly of complex genomic regions that are difficult to resolve using short-read sequencing technologies. Additionally, they facilitate the identification of structural variations at high resolution, helping researchers understand their impact on genetic diversity and phenotypic traits.
By investigating the pan-genome and its structural variations, researchers aim to uncover the genetic basis of various traits and understand the mechanisms underlying adaptation, evolution, and disease susceptibility in plant and animal populations. This knowledge can have practical applications in areas such as crop improvement, breeding programs, and personalized medicine.
Crop Pan-Genome Material Selection
Number of Materials
One of the key determinants of pan-genome size is the percentage of non-core genes, which can range from 8% to 61% in crop pan-genome studies. Sample size plays a vital role in such studies. Initially, as the number of individuals with newly identified genes increases, the pan-genome expands. However, this expansion is accompanied by a decrease in the proportion of core genes.
Characteristics of Materials
The selection of materials has a profound influence on the efficiency and integrity of pan-genome studies. Two crucial characteristics warrant attention: (1) Proximity of Relatives: Choosing closely related materials tends to underestimate the size of the pan-genome. Therefore, it is important to include a diverse range of individuals to obtain a comprehensive understanding of the crop's genetic landscape. (2) Combination of Wild and Cultivated Germplasm: The combination of wild and cultivated germplasm results in a larger species-size pan-genome, with a significantly higher proportion of non-core genes compared to using cultivated germplasm alone. Incorporating wild materials enhances the diversity and inclusiveness of the pan-genome.
In crop research, the identification of new genes tends to decrease as the number of sequenced study materials increases. This suggests that there is a finite number of genomes beyond which additional inclusion does not lead to further pan-genome expansion. Moreover, during crop domestication, the lack of genetic diversity negatively affects the size of the pan-genome and the proportion of non-core genes. Increasing the inclusion of wild materials can help mitigate this issue by elevating the percentage of core genes in the pan-genome. Crops with limited reduction in diversity during domestication tend to exhibit a higher proportion of non-core genes. The proportion of non-core genes is an indicator of species diversity and can be influenced by factors such as ploidy level, reproduction method, and bottleneck periods during domestication. Higher ploidy levels and heterozygous hybridization rates contribute to increased diversity and tolerance to deleterious mutations, resulting in a pan-genome with a higher percentage of non-core genes.
Constructing the Pan-genome
The construction of a pan-genome revolves around identifying the variations in gene presence or absence among individuals. This entails segregating similar sequences into distinct alleles, extra copies, or non-essential genes. The challenge lies in the difficulty of discerning inter-individual variations due to the sequence similarities. Therefore, gathering information on the physical location and gene order in the assembled genome becomes crucial. There are three primary methods employed for constructing a pan-genome: Iterative, map-to-pan, and De novo assembly.
The Iterative and map-to-pan methods involve identifying presence/absence variations (PAVs) of genes by comparing short reads to the annotated genome. Conversely, the De novo assembly method is utilized to further infer PAVs of genes by comparing the assembled genes with the annotated ones. As a result, this method provides more accurate information about the pan-genome. However, achieving high assembly quality genomes through De novo assembly requires a high sequencing depth, which comes at a significant cost.
On the other hand, iterative assembly and map-to-pan techniques allow pan-genome studies to be conducted at relatively low sequencing depths, thereby reducing costs and enabling a larger pool of individual samples for selection. In addition to the assembly method, the number of individuals and the genetic relationships among them play a crucial role in ensuring the comprehensiveness of pan-genome studies. They also determine the accuracy of estimating the pan-genome's size.
Advancements in sequencing technologies, especially long-read sequencing techniques and assembly methods, have significantly lowered the cost of achieving high-quality de novo assembly. This, in turn, will facilitate future studies employing de novo assembly methods.
Reference:
- Khan, Aamir W., et al. "Super-pangenome by integrating the wild side of a species for accelerated crop improvement." Trends in Plant Science 25.2 (2020): 148-158.


