
 Sample Submission Guidelines
Sample Submission Guidelines
Pan-Genome Analysis Tools: A Comprehensive Overview
As a transformative approach in modern genomics, pan-genome analysis investigates the collective genetic content across all members of a species. Cutting-edge sequencing technologies now permit the simultaneous examination of numerous genomes, elucidating both essential core sequences and strain-specific genetic components. Such analyses provide unprecedented resolution in mapping genotype-phenotype relationships at population scales. The formidable data processing requirements have catalyzed innovation in specialized analytical pipelines. Here, we survey principal computational tools that facilitate pan-genome reconstruction and biological interpretation in contemporary genomic research.
Services you may interested in
Learn More
Pan-Genome Construction Tools
In the era of genomics, the construction of pan-genomes has become a crucial approach to capturing the genetic diversity within and across species. Pan-genome analysis allows us to identify core and accessory genes, understand evolutionary dynamics, and explore the functional variations that drive biological traits. The process involves several key steps: genome assembly using tools like SPAdes or Flye, annotation with software such as Prokka or RAST, and the identification of orthologous gene clusters through OrthoFinder or Roary. Comparative analysis is facilitated by phylogenetic tools like RAxML and variation analysis tools like Snippy. Visualization and interpretation are supported by Circos and statistical analysis with R or Python. These tools are essential for handling the complexities of genomic data, from sequence assembly and annotation to the integration and comparison of multiple genomes. By leveraging these tools, researchers can effectively build comprehensive pan-genomes that provide deep insights into the genetic architecture of organisms and their populations.
Panaroo
Building on the essential toolkit for pan-genome construction, we now delve into a specific yet powerful component: Panaroo. This section focuses on how Panaroo processes multiple bacterial genomes to construct a comprehensive pan-genome. By identifying core and accessory genes through advanced clustering and alignment techniques, Panaroo generates a gene presence/absence matrix. This matrix is crucial for downstream analyses, providing valuable insights into genetic diversity, phylogenetics, and functional gene distribution.
Functionality: Panaroo uses a combination of clustering algorithms and sequence alignment methods to identify homologous genes across multiple genomes. It constructs a gene presence/absence matrix, which is essential for downstream analyses such as phylogenetic tree construction and gene frequency calculations.
Applications: Panaroo is widely used in microbiology and infectious disease research. It helps understand bacterial populations' genetic diversity and identify virulence and antibiotic-resistance genes.
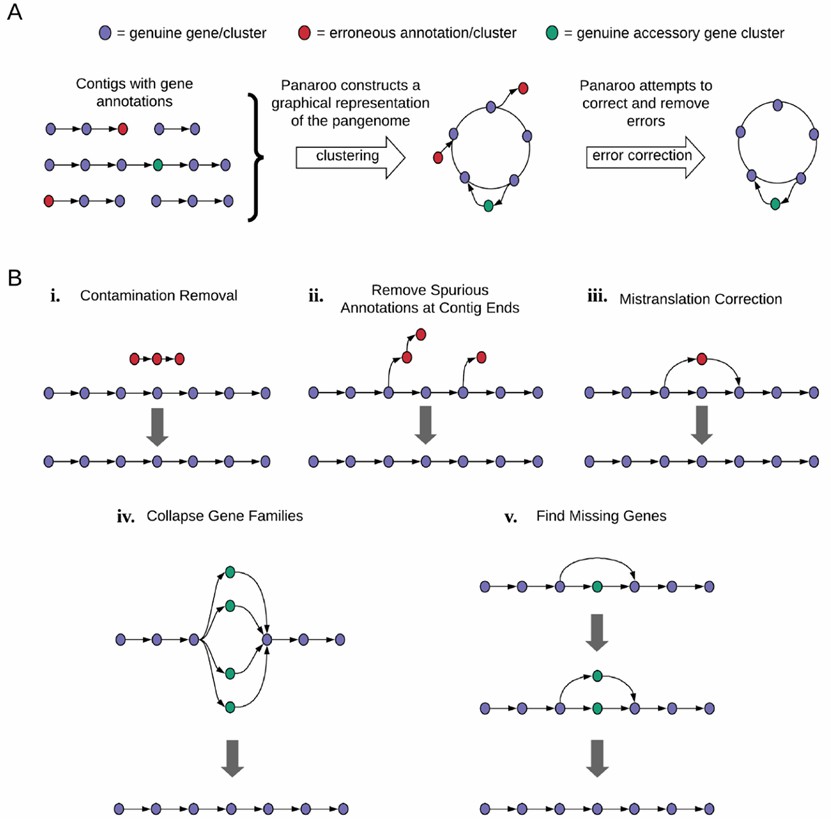 Figure 1. Panaroo is used to correct annotation errors.(Gerry Tonkin-Hill, et. al,2020)
Figure 1. Panaroo is used to correct annotation errors.(Gerry Tonkin-Hill, et. al,2020)
In bacterial diversity studies, Panaroo can be used to analyze the core and accessory genes of a large number of strains, helping scientists understand the molecular basis of species adaptability and evolution. For example, researchers can compare samples of Escherichia coli from different environments and use Panaroo to identify sets of genes associated with specific environmental adaptations.
Roary
Following the introduction of essential pan-genome construction tools, we now delve into a specific yet powerful tool in this domain: Roary. By using Roary, researchers can obtain detailed data on gene presence/absence patterns, core and accessory genome sizes, which are crucial for understanding bacterial evolution and comparative genomics.
Functionality: Roary clusters orthologous genes using the OrthoFinder algorithm and constructs a pan-genome matrix. It also provides detailed reports on gene presence/absence, core genome size, and accessory genome size.
Applications: Roary is extensively used in bacterial genomics to study the evolution of bacterial pathogens and the spread of antibiotic-resistant genes. It is also helpful for comparative genomics studies across different bacterial species.
PanX
PanX is a versatile tool designed for constructing and analyzing pan-genomes of bacterial and viral species. It identifies orthologous genes using BLAST and MCL clustering, constructs pan-genome graphs, and visualizes gene presence/absence patterns. Additionally, PanX detects SNPs and indels, providing valuable insights into genetic diversity and evolutionary dynamics.
Functionality: PanX uses a combination of BLAST and MCL clustering algorithms to identify orthologous genes. It constructs pan-genome graphs and provides detailed visualizations of gene presence/absence patterns. PanX also supports the identification of single nucleotide polymorphisms (SNPs) and insertion/deletion events.
Applications: PanX is used in various microbiological studies, including the analysis of bacterial and viral pan-genomes. It is particularly useful for researchers who need a comprehensive and integrated platform for pan-genome analysis.
PanGP
PanGP is a powerful tool for constructing pan-genomes and identifying core and accessory genes. It analyzes multiple genomes within a species to provide insights into genetic diversity. By using sequence alignment and clustering algorithms, PanGP identifies orthologous genes and constructs a pan-genome matrix. This tool offers detailed reports on gene presence/absence patterns and supports the identification of gene family expansions and contractions. Researchers can use PanGP to gain a comprehensive understanding of the genetic architecture and evolutionary dynamics within a species.
Functionality: PanGP uses a combination of sequence alignment and clustering algorithms to identify orthologous genes. It constructs a pan-genome matrix and provides detailed reports on gene presence/absence patterns. PanGP also supports the identification of gene family expansions and contractions.
Applications: PanGP is used in various genomics studies, including the analysis of bacterial and plant pan-genomes. It is particularly useful for researchers who need to identify core and accessory genes and understand the genetic diversity within a species.
Variant Calling and Genotyping Tools
Snippy
Snippy is a powerful bioinformatics tool designed to analyze next-generation sequencing data. It aligns reads to a reference genome and identifies variants such as SNPs and indels. This process generates detailed variant data, including frequencies and genotypes, which are crucial for understanding genetic diversity and evolutionary relationships.
Functionality: Snippy aligns sequencing reads to a reference genome and identifies SNPs, indels, and other variants. It uses a combination of mapping and assembly-based approaches to improve the accuracy of variant calling. Snippy also provides detailed reports on variant frequencies and genotypes.
Applications: Snippy is widely used in bacterial genomics for strain typing, phylogenetic analysis, and the study of genetic diversity. It is particularly useful for researchers working with large datasets that require rapid and accurate variant calling.
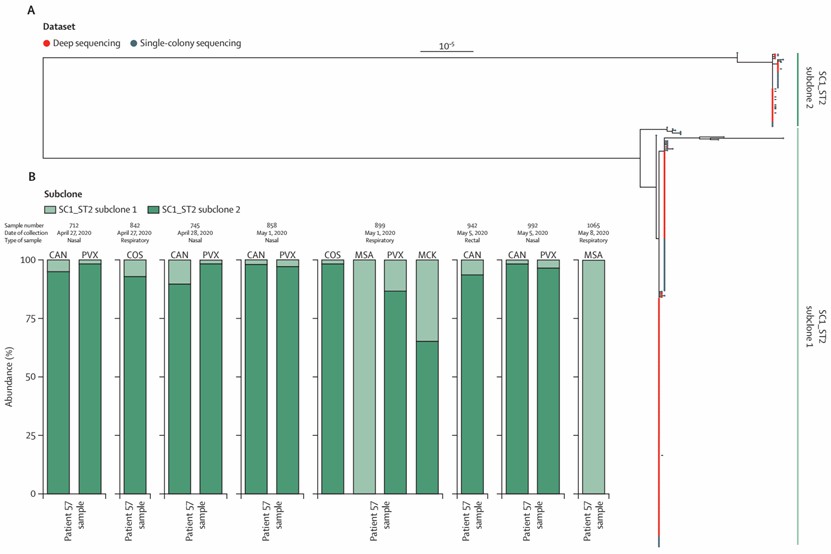 Figure 2. Reads were mapped to a common reference (SAMN07258611) by the use of Snippy.(Thorpe, et. al,2024)
Figure 2. Reads were mapped to a common reference (SAMN07258611) by the use of Snippy.(Thorpe, et. al,2024)
GATK
In this section, we delve into the GATK, a powerful suite of tools designed for variant discovery and genotyping in high-throughput sequencing data. GATK is instrumental in identifying genetic variations such as SNPs and insertions/deletions with high accuracy. By leveraging Bayesian statistical models, it enhances the reliability of variant detection and genotyping. Researchers can obtain detailed variant data, which is crucial for understanding genetic diversity, disease associations, and evolutionary processes.
Functionality: GATK includes a range of tools for variant calling, including HaplotypeCaller and GenotypeGVCFs. It uses Bayesian statistical models to improve the accuracy of variant detection and genotyping. GATK also provides tools for variant filtering and annotation.
Applications: GATK is used in various genomics studies, including human disease research, plant breeding, and population genomics. It is particularly useful for researchers who require high accuracy and reproducibility in variant calling and genotyping.
FreeBayes
In the realm of genomics, identifying genetic variations is crucial for understanding biological diversity and function. FreeBayes, an open-source variant calling tool, excels in this task. It processes next-generation sequencing data to detect variants, including SNPs and indels, providing detailed quality metrics. FreeBayes is versatile, supporting both diploid and polyploid genomes, and is widely used in human disease research, plant breeding, and population genomics.
Functionality: FreeBayes uses a Bayesian framework to call variants from sequencing reads. It supports diploid and polyploid genomes and can handle complex types such as structural and copy number variations. FreeBayes also provides detailed reports on variant quality metrics and genotypes.
Applications: FreeBayes is used in various genomics studies, including human disease research, plant breeding, and population genomics. It is particularly useful for researchers who require accurate and flexible variant calling from diverse datasets.
Visualization Tools
DeepVariant
DeepVariant is a powerful tool designed to accurately detect genetic variants from sequencing data. It leverages deep learning to analyze aligned reads and identify SNPs and indels. By using convolutional neural networks, DeepVariant enhances variant detection accuracy, providing reliable data for downstream analysis. This tool is essential for researchers aiming to achieve high-quality variant calling in diverse genomic studies.
Functionality: DeepVariant takes aligned sequencing reads as input and uses a deep-learning model to call variants. It supports both SNPs and indels and provides detailed reports on variant quality metrics and genotypes. DeepVariant also integrates with other tools, such as GATK, for variant filtering and annotation.
Applications: DeepVariant is used in various genomics studies, including human disease research and population genomics. It is particularly useful for researchers who require high accuracy and reproducibility in variant calling.
Comparative Genomics Tools
OrthoFinder
OrthoFinder is a powerful tool for comparative genomics, designed to identify orthologous genes and construct phylogenetic trees. It is widely used in both bacterial and plant genomics. This tool helps researchers analyze gene families across multiple genomes, providing insights into evolutionary relationships and functional variations. By identifying orthologous genes and constructing phylogenetic trees, OrthoFinder offers detailed reports on gene family expansions and contractions, which are crucial for understanding the genetic diversity and evolutionary dynamics of species.
Functionality: OrthoFinder uses a combination of sequence alignment and clustering algorithms to identify orthologous genes across multiple genomes. It constructs phylogenetic trees based on gene presence/absence data and provides detailed reports on gene family expansions and contractions.
Applications: OrthoFinder is used in various comparative genomics studies, including the analysis of bacterial and plant pan-genomes. It is particularly useful for researchers who need to identify orthologous genes and understand the evolutionary relationships among species.
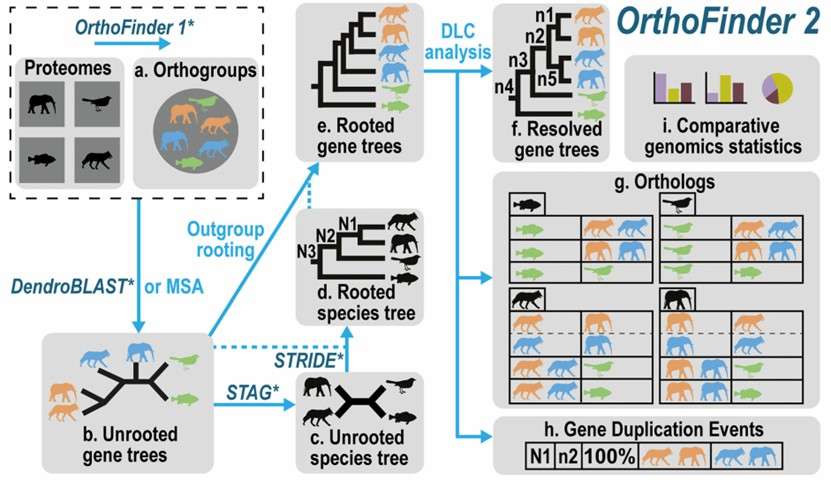 Figure 3. The OrthoFinder workflow. (Emms, et. al,2019)
Figure 3. The OrthoFinder workflow. (Emms, et. al,2019)
Anvi'o
Anvi'o is a powerful tool designed for the analysis and visualization of microbial genomes. It integrates multiple functionalities to support genome assembly, variant calling, and comparative analysis. By using Anvi'o, researchers can construct phylogenetic trees, generate gene presence/absence matrices, and create detailed visualizations such as heat maps and circular plots. This platform is particularly useful for studying bacterial and viral genomes, providing valuable insights into microbial diversity and evolution.
Functionality: Anvi'o includes tools for genome assembly, variant calling, and comparative analysis. It supports the construction of phylogenetic trees, gene presence/absence matrices, and pan-genome graphs. Anvi'o also provides detailed visualizations of genomic data, including heat maps and circular plots.
Applications: Anvi'o is used in various microbiological studies, including the analysis of bacterial and viral genomes. It is particularly useful for researchers who need a comprehensive and integrated platform for comparative genomics.
PanPhlan
PanPhlan is a powerful comparative genomics tool specifically designed for analyzing the pan-genomes of microbial communities. This tool is essential for metagenomic studies, allowing researchers to identify and compare orthologous genes across multiple genomes. By constructing pan-genome graphs and generating detailed reports on gene presence/absence patterns, PanPhlan provides valuable insights into the functional diversity of microbial communities. It is particularly useful for comparing pan-genomes from different environments or conditions, helping researchers understand the genetic variations and evolutionary dynamics within microbial populations.
Functionality: PanPhlan uses a combination of sequence alignment and clustering algorithms to identify orthologous genes across multiple genomes. It constructs pan-genome graphs and provides detailed reports on gene presence/absence patterns. PanPhlan also supports the comparison of pan-genomes from different microbial communities.
Applications: PanPhlan is used in various microbiological studies, including the analysis of bacterial and viral pan-genomes. It is particularly useful for researchers who need to compare the pan-genomes of different microbial communities and understand their functional diversity.
Conclusion
The emergence of pan-genome analytics has fundamentally transformed contemporary genomic research, enabling comprehensive characterization of intra-species variation and evolutionary patterns. This shift from traditional single-genome analysis to a pan-genomic perspective allows researchers to capture the full spectrum of genetic diversity within and across species, revealing both core and accessory genomic elements. The ability to identify and compare these elements provides critical insights into the functional and evolutionary dynamics that shape microbial communities and other biological systems.
Modern computational pipelines now facilitate high-precision assembly, interrogation, and graphical representation of pan-genomic data through specialized platforms. For genome construction, tools like Panaroo and Roary have become indispensable, allowing for the efficient identification of orthologous gene clusters and the construction of pan-genome matrices. These matrices serve as the foundation for understanding the presence and absence of genes across multiple genomes, highlighting the core genes that are essential for survival and the accessory genes that contribute to niche adaptation and functional diversity.
Variant detection has also seen significant advancements, with tools like Snippy and GATK enabling the identification of SNPs and other genetic variations. These variations are crucial for understanding the evolutionary relationships between different strains or species and for tracing the spread of specific traits or diseases. The ability to detect and analyze these variants at a high resolution has greatly enhanced our understanding of microbial evolution and adaptation.
Visualization tools such as PanX and Circos play a vital role in making complex pan-genomic data more accessible and interpretable. These tools provide intuitive graphical representations of pan-genome data, allowing researchers to visualize gene presence/absence patterns, phylogenetic relationships, and other key features. By presenting data in a visually compelling format, these tools facilitate the identification of trends and patterns that might otherwise go unnoticed.
Furthermore, advanced comparative frameworks including OrthoFinder, Anvi'o, and PanPhlan enable cross-taxa genomic investigations, particularly in microbial systems. These tools allow researchers to compare pan-genomes from different microbial communities, providing insights into the functional diversity and evolutionary dynamics across a wide range of organisms. By integrating data from multiple sources and employing sophisticated algorithms, these frameworks enable researchers to uncover the genetic basis of ecological interactions, host-pathogen relationships, and other critical biological phenomena.
In summary, the advent of pan-genome analytics has revolutionized genomic research by providing a more comprehensive and nuanced understanding of genetic diversity. Modern computational tools and pipelines have made it possible to assemble, analyze, and visualize pan-genomic data with unprecedented precision. These advances are proving indispensable for deciphering the biological implications of genetic diversity across different organisms, ultimately enhancing our ability to address fundamental questions in biology, ecology, and medicine.
References:
- Tonkin-Hill, G., MacAlasdair, N., et, al. (2020). Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome biology, 21(1), 180. https://doi.org/10.1186/s13059-020-02090-4
- Thorpe, H. A., Pesonen, M., et, al. (2024). Pan-pathogen deep sequencing of nosocomial bacterial pathogens in Italy in spring 2020: a prospective cohort study. The Lancet. Microbe, 5(10), 100890. https://doi.org/10.1016/S2666-5247(24)00113-7
- Emms, D. M., & Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome biology, 20(1), 238. https://doi.org/10.1186/s13059-019-1832-y


