
 Sample Submission Guidelines
Sample Submission Guidelines
Comprehensive Overview of Chip-seq
In the field of epigenetics, chromatin immunoprecipitation (ChIP) serves as a fundamental methodology, facilitating a nuanced investigation into the intricate interplay between proteins and DNA within the chromatin milieu. Among its diverse methodologies, immunoprecipitation followed by sequencing (ChIP-seq) has arisen as a potent instrument, providing unprecedented elucidation into genome-wide protein-DNA interactions. Within this article, we meticulously dissect the nuances of ChIP-seq, delineating its applications, mechanisms, and merits, thereby illuminating its pivotal contribution to the elucidation of chromatin biology's enigmas.
What is Chromatin Immunoprecipitation?
Chromatin, a dynamic amalgamation of DNA and its associated proteins, assumes a critical role in the governance of gene expression and genome architecture. Within this complex framework, proteins including transcription factors, histones, and chromatin modifiers intricately choreograph DNA accessibility and transcriptional dynamics. ChIP represents a methodological approach meticulously crafted to unveil the precise binding loci of these proteins across the genome.
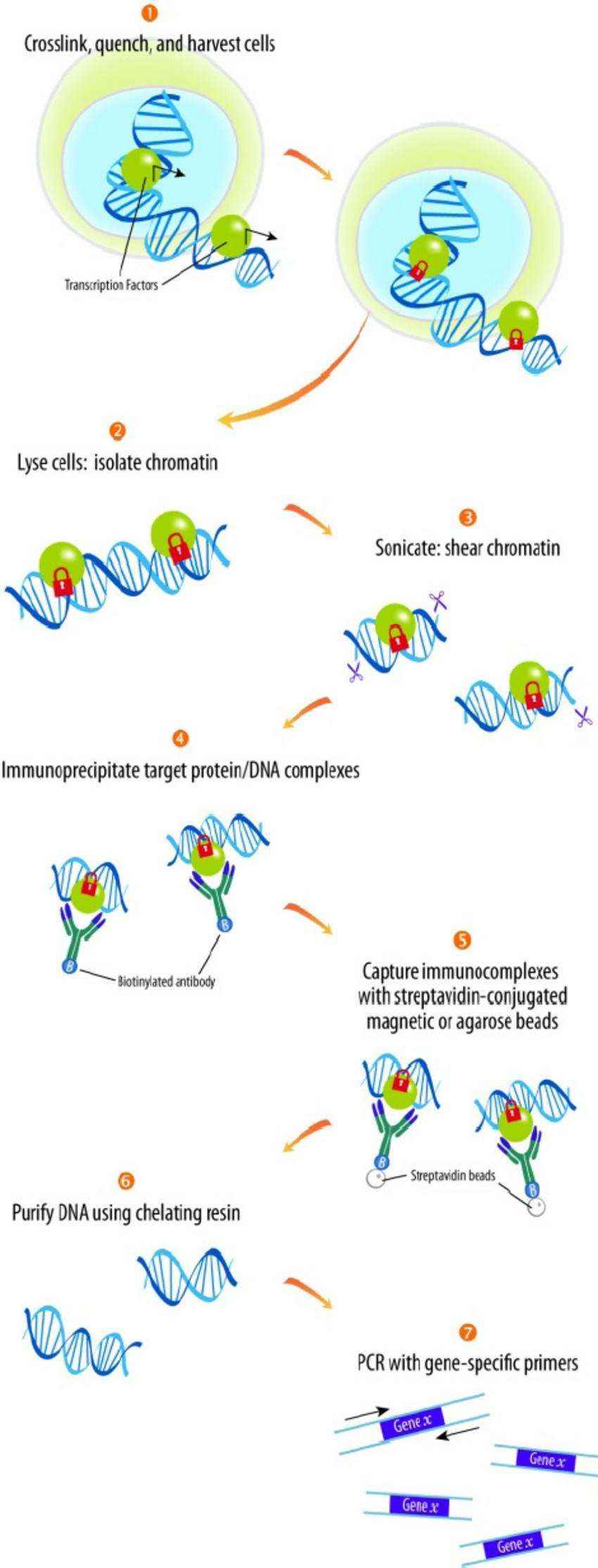 Principle of ChIP technique (Alain Rival et al, 2010)
Principle of ChIP technique (Alain Rival et al, 2010)
What is ChIP-seq?
ChIP-seq epitomizes a robust molecular biology methodology engineered to delineate the precise binding sites of proteins across the chromatin milieu with unparalleled precision and genomic breadth. The foundational principle underpinning ChIP-seq centers on the targeted enrichment of DNA fragments tethered to proteins of interest, succeeded by high-throughput sequencing endeavors aimed at cartographing these interactions across the entirety of the genome.
You may interested in
ChIP-seq Application: Illuminating Epigenetic Landscapes
Deciphering Developmental Processes
ChIP-seq has catalyzed a paradigm shift in our comprehension of developmental biology, unveiling the intricate regulatory networks governing embryogenesis and tissue differentiation. For example, Bernstein et al. (2006) conducted a seminal investigation employing ChIP-seq to map histone modifications during mouse embryonic stem cell differentiation, thereby elucidating dynamic alterations in chromatin structure linked to lineage specification (Bernstein et al., 2006). Moreover, recent advancements in single-cell ChIP-seq methodologies have empowered the interrogation of epigenetic dynamics with unprecedented resolution, enabling researchers to delineate the chromatin landscapes of individual cells during developmental transitions (Rotem et al., 2015). Consequently, ChIP-seq stands as a foundational technology pivotal in unraveling the epigenetic mechanisms governing cell fate determination and organogenesis.
Unraveling Cancer Epigenetics
In the domain of cancer research, ChIP-seq emerges as a pivotal tool, offering profound insights into the epigenetic modifications steering tumorigenesis and the trajectory of disease progression. A notable illustration of its utility is evidenced in a seminal work by Dawson et al. (2012), wherein ChIP-seq was deployed to delineate histone modifications within prostate cancer cells, thereby discerning discrete chromatin signatures intricately linked to oncogenic gene expression paradigms (Dawson et al., 2012). Furthermore, through integrative ChIP-seq analyses, researchers have elucidated the intricate interplay between transcription factor binding and chromatin accessibility in cancerous cells, thereby shedding light on the multifaceted regulatory cascades underpinning malignant metamorphosis (Jin et al., 2015). By harnessing the formidable capabilities of ChIP-seq, scholars are empowered to decode the epigenomic terrain of cancerous genomes and pinpoint hitherto undiscovered therapeutic targets, thus propelling the advancement of precision oncology interventions.
Mapping Regulatory Elements
In addition to its pivotal roles in developmental and cancer biology, ChIP-seq serves as a cornerstone for the systematic mapping of regulatory elements, including enhancers, promoters, and insulators, throughout the genome. Noteworthy is a seminal investigation by Heinz et al. (2010), which leveraged ChIP-seq to meticulously annotate enhancer elements and transcription factor binding sites within human CD4+ T cells, thereby unveiling intricate cell type-specific regulatory networks pivotal in orchestrating immune responses (Heinz et al., 2010). Furthermore, ChIP-seq has proven indispensable in unraveling the epigenetic orchestration governing non-coding RNA species, thereby providing invaluable insights into their regulatory roles in gene expression dynamics and cellular equilibrium (Marson et al., 2008). By illuminating the regulatory blueprint of the genome, ChIP-seq catalyzes the identification of pivotal genomic elements dictating gene expression trajectories in both physiological states and pathological conditions.
Embryonic Stem Cell Pluripotency and Differentiation
Investigations harnessing ChIP-seq have played a pivotal role in advancing our comprehension of embryonic stem cell (ESC) pluripotency and the intricacies of cellular differentiation. A seminal work by Bernstein et al. (2006) stands out, wherein ChIP-seq was employed to elucidate bivalent chromatin domains characterized by the concurrent presence of activating (H3K4me3) and repressive (H3K27me3) histone modifications at critical developmental loci within ESCs. This delineation underscored the poised nature of these genes, primed for either activation or repression as cells undergo lineage-specific differentiation. Such groundbreaking endeavors have furnished invaluable insights into the epigenetic machinery governing pluripotency and the pivotal transition towards lineage commitment.
Cell Subpopulation Identification
Furthermore, the advent of single-cell ChIP-seq (scChIP-seq) has revolutionized our ability to delineate discrete cell subpopulations by their chromatin landscapes. A pivotal demonstration of this capability was showcased by Rotem et al. (2015), who underscored the efficacy of scChIP-seq in pinpointing cell subsets characterized by distinct chromatin signatures. Their work not only unveiled the intricate heterogeneity inherent within cellular populations but also unveiled the regulatory frameworks orchestrating such diversity. By meticulously profiling individual cells at unparalleled resolution, scChIP-seq emerges as a potent tool, offering unprecedented glimpses into the epigenetic intricacies governing cellular heterogeneity throughout developmental trajectories.
Epigenetic Regulation of Disease States
Additionally, ChIP-seq investigations have provided insights into the epigenetic modifications linked with diverse pathological conditions, thereby identifying promising avenues for therapeutic intervention. In a seminal work by Dawson et al. (2012), ChIP-seq was deployed to scrutinize the involvement of bromodomain and extraterminal (BET) proteins in MLL-fusion leukemia, unveiling the therapeutic promise of BET inhibition in mitigating oncogenic chromatin configurations and curtailing leukemic progression. This investigation serves as a paradigm, showcasing the capacity of ChIP-seq to unveil disease-specific epigenetic profiles and contribute substantively to the refinement of targeted therapeutic modalities.
Three-Dimensional Chromatin Organization
ChIP-seq has emerged as a pivotal tool in unraveling the three-dimensional (3D) architecture of chromatin and its implications for gene regulation. Jin et al. (2013) harnessed ChIP-seq in conjunction with chromosome conformation capture (3C) methodologies to construct a finely detailed map of the chromatin interactome within human cells, thereby unveiling intricate spatial relationships between distal regulatory elements and their cognate target genes. This integrative strategy affords valuable perspectives into the spatial deployment of regulatory elements and their pivotal roles in orchestrating developmental gene expression programs.
Lineage-Determining Transcription Factors
Moreover, ChIP-seq inquiries have shed light on the pivotal function of lineage-determining transcription factors in governing cellular fate determination and lineage commitment. Heinz et al. (2010) employed ChIP-seq to delineate lineage-specific cis-regulatory modules governed by pivotal transcription factors like PU.1 and C/EBP during macrophage and B cell differentiation. Through the delineation of binding sites orchestrated by lineage-specifying transcription factors, ChIP-seq unveils mechanistic insights into the regulatory cascades underpinning cellular fate specification.
MicroRNA-Mediated Regulation
Moreover, ChIP-seq has been instrumental in deciphering the regulatory circuitry of microRNA-mediated gene regulation during development. Marson et al. (2008) utilized ChIP-seq to connect microRNA genes to the core transcriptional regulatory circuitry of ESCs, revealing the interplay between transcription factors and microRNAs in controlling pluripotency and differentiation. This integrative approach elucidates the epigenetic mechanisms underlying microRNA-mediated gene regulation and their impact on developmental processes.
How Does ChIP-seq Work?
Traditionally, ChIP-chip, employing microarray technology, was utilized for mapping protein-DNA interactions. However, the advent of ChIP-seq supplanted ChIP-chip owing to its higher resolution, dynamic range, and genome-wide coverage. In ChIP-chip, the immunoprecipitated DNA fragments are hybridized to microarrays containing probes complementary to specific genomic regions, allowing for the identification of bound sites.
On the contrary, ChIP-seq utilizes advanced sequencing techniques to analyze the enriched DNA fragments, facilitating the precise localization of protein-binding events across the entirety of the genome. After the immunoprecipitation of protein-DNA complexes, the DNA fragments in question undergo sequencing, yielding millions of short reads which are then aligned to the reference genome. The resultant profile of read density offers insights into the distribution of protein binding sites, thus furnishing a comprehensive map of chromatin occupancy.
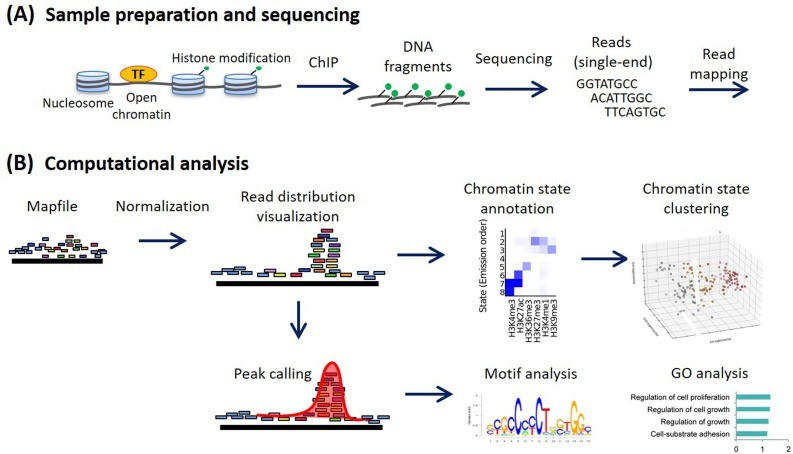 ChIP-seq analysis workflow
ChIP-seq analysis workflow
Advantages of ChIP-seq
The transition from ChIP-chip to ChIP-seq heralded several advantages, catapulting chromatin biology into a new era of discovery.
High Resolution: ChIP-seq offers base-pair resolution, allowing for precise localization of protein-binding sites and identification of narrow peaks, which is particularly advantageous for studying transcription factor binding events and histone modifications.
Global Coverage: Unlike ChIP-chip, which relies on predefined microarray probes, ChIP-seq provides genome-wide coverage, enabling the identification of novel binding sites and regulatory elements without prior knowledge.
Dynamic Range: ChIP-seq exhibits a broader dynamic range and increased sensitivity compared to ChIP-chip, enabling detection of low-abundance protein-DNA interactions and subtle changes in chromatin states.
Data Reproducibility: The reproducibility of ChIP-seq data is enhanced due to its digital nature, minimizing technical variability and facilitating robust comparison between samples.
Flexibility: ChIP-seq is amenable to various sample types, including limited biological materials such as clinical specimens and single cells, broadening its applicability in translational and clinical research.
Learn More
Chip-seq Illumina
ChIP-seq technology, particularly when utilizing Illumina platforms, signifies a transformative advancement in epigenomic exploration, characterized by its remarkable resolution and sensitivity in elucidating DNA-protein interactions and histone modifications. Leveraging cutting-edge sequencing platforms from Illumina, this methodology facilitates the exhaustive mapping of chromatin modifications and transcription factor binding sites throughout the genome with exceptional precision and efficiency. Through the integration of chromatin immunoprecipitation with next-generation sequencing, ChIP-seq technology utilizing Illumina instrumentation has emerged as a formidable instrument for unraveling the intricate epigenetic landscapes that govern gene expression, developmental processes, and pathological conditions.
Advantages of Chip-seq Illumina
ChIP-seq using Illumina technology offers numerous advantages over conventional sequencing methodologies, encompassing heightened throughput, diminished sequencing expenditures, and augmented sensitivity. Leveraging Illumina's state-of-the-art sequencing platforms facilitates the acquisition of top-tier data quality even with limited input material, rendering it particularly adept for probing rare cell populations and invaluable clinical specimens. Furthermore, the scalability inherent to Illumina sequencing permits sample multiplexing, thereby empowering expansive epigenomic investigations and hastening breakthroughs in chromatin biology.
Advanced Analysis of Chip-seq Data
Bioinformatics Pipelines for Chip-seq Analysis
Examining ChIP-seq data demands resilient bioinformatics pipelines adept at processing raw sequencing reads, discerning enriched regions, and annotating regulatory elements with precision and reproducibility. Sophisticated algorithms and stringent quality control measures are deployed to ensure the dependability of results in Chip-seq analysis. Bioinformatics specialists adopt a thorough methodology for data analysis, which encompasses read alignment, peak calling, differential binding analysis, motif discovery, and pathway enrichment analysis. The amalgamation of diverse layers of epigenomic data yields valuable insights into gene regulatory networks and chromatin dynamics, thereby facilitating the exploration of the intricacies of genome regulation.
Quality Control and Data Visualization
Quality assurance holds paramount importance in the analysis of Chip-seq data, ensuring both the accuracy and reliability of the data under scrutiny. Rigorous quality control protocols are meticulously executed to evaluate various facets of data quality, encompassing assessments of sequencing depth, read distribution, and artifact identification. Leveraging advanced data visualization tools alongside interactive plots facilitates comprehensive exploration and interpretation of the data. Heatmaps, metaplots, genome browser tracks, and differential binding profiles serve as invaluable aids in intuitively grasping the spatial distribution of regulatory elements and epigenetic modifications across the genome landscape.
Learn More
ChIP-seq vs. RNA-seq
Comparative analyses between ChIP-seq and RNA-seq underscore the synergistic relationship between these two pivotal sequencing methodologies in elucidating gene regulatory networks. ChIP-seq unveils the spatial arrangements of DNA-binding proteins and histone modifications, furnishing invaluable insights into the chromatin landscape. Conversely, RNA-seq delivers quantitative evaluations of gene expression profiles and transcriptomic fluctuations, illuminating the dynamic nature of cellular processes.
The amalgamation of ChIP-seq and RNA-seq datasets yields a panoramic depiction of gene regulatory modalities, encapsulating pivotal events ranging from chromatin accessibility and transcription factor interactions to mRNA synthesis and post-transcriptional modifications. This integrative approach affords a profound comprehension of cellular physiology and pathological mechanisms, fostering advancements in our understanding of intricate biological phenomena and disease etiology.
ChIP-seq vs. ATAC-seq
ChIP-seq and assay for transposase-accessible chromatin followed by sequencing (ATAC-seq) represent complementary methodologies within the realm of chromatin biology. ChIP-seq enables the generation of intricate maps depicting protein-DNA interactions and histone modifications, facilitating the precise interrogation of predetermined genomic loci. In stark contrast, ATAC-seq affords a panoramic vantage of chromatin accessibility across the entire genome, identifying regulatory elements devoid of prior knowledge regarding specific protein binding sites. While the application of ChIP-seq necessitates larger quantities of cellular material and furnishes focused insights, ATAC-seq thrives on smaller cell populations, affording expansive coverage of genomic landscapes. The integration of data gleaned from both methodologies empowers researchers to unravel the intricate interplay between chromatin accessibility and the dynamics of protein binding, thereby furnishing a holistic understanding of chromatin's structural and functional intricacies.
You may interested in
Learn More
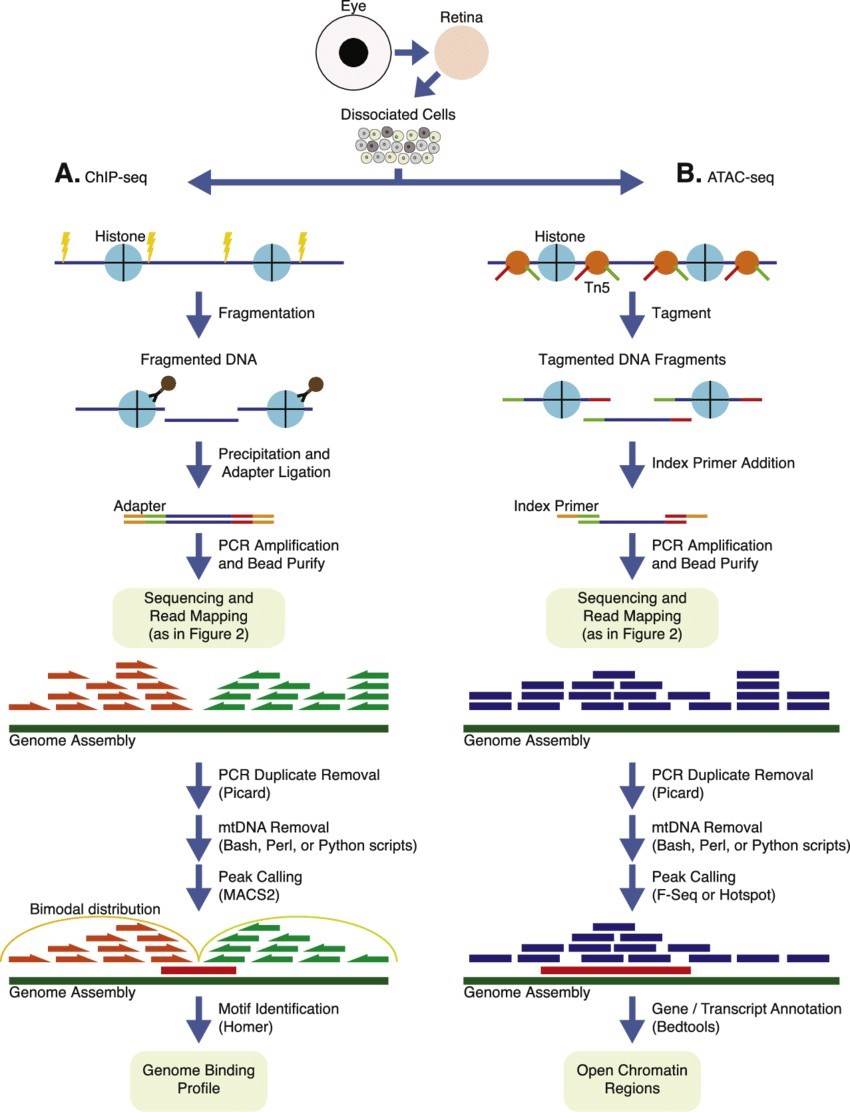 ChIP and ATAC sequencing workflow and analysis. (Vijender Chaitankar et al, 2016)
ChIP and ATAC sequencing workflow and analysis. (Vijender Chaitankar et al, 2016)
ChIP-seq vs. Cut-and-Run
Though ChIP-seq has long served as the benchmark method for delineating protein-DNA interactions, the emergence of alternative approaches, such as Cut-and-Run, presents promising alternatives. Cut-and-Run employs antibody-directed cleavage of protein-DNA complexes, followed by high-throughput sequencing, boasting advantages in terms of simplicity, sensitivity, and specificity over traditional ChIP-seq methodologies. Nonetheless, each technique harbors distinct strengths and limitations, and the selection between them hinges upon the precise experimental prerequisites and biological inquiries at hand.
Learn More
Summary
The article delves into the pivotal role of ChIP-seq in unraveling the complexities of chromatin biology. Through meticulous exploration, it highlights ChIP-seq's applications, mechanisms, and merits, showcasing its indispensable contributions to our understanding of epigenetics. From deciphering developmental processes to elucidating cancer epigenetics and mapping regulatory elements, ChIP-seq emerges as a transformative tool, offering unprecedented insights into gene regulation, cellular differentiation, and disease mechanisms. Its integration with other advanced methodologies promises to deepen our comprehension of the epigenomic landscape, paving the way for innovative therapeutic interventions and further advancements in chromatin biology.
References:
- Bernstein, B. E., Mikkelsen, T. S., Xie, X., Kamal, M., Huebert, D. J., Cuff, J., ... & Lander, E. S. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell, 125(2), 315-326.
- Rotem, A., Ram, O., Shoresh, N., Sperling, R. A., Goren, A., Weitz, D. A., & Bernstein, B. E. (2015). Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nature biotechnology, 33(11), 1165-1172.
- Dawson, M. A., Prinjha, R. K., Dittmann, A., Giotopoulos, G., Bantscheff, M., Chan, W. I., ... & Knapp, S. (2012). Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature, 478(7370), 529-533.
- Jin, F., Li, Y., Dixon, J. R., Selvaraj, S., Ye, Z., Lee, A. Y., ... & Ren, B. (2013). A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature, 503(7475), 290-294.
- Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C., Laslo, P., ... & Glass, C. K. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell, 38(4), 576-589.
- Marson, A., Levine, S. S., Cole, M. F., Frampton, G. M., Brambrink, T., Johnstone, S., ... & Young, R. A. (2008). Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell, 134(3), 521-533.
- Ryuichiro Nakato, Toyonori Sakata. Methods for ChIP-seq analysis: A practical workflow and advanced applications. Methods, 2021


