
 Sample Submission Guidelines
Sample Submission Guidelines
ChIP-seq vs. CUT&RUN
In contemporary molecular biology research, the investigation of protein-DNA interactions and epigenetic modifications stands as a cornerstone for understanding chromatin dynamics and its impact on gene expression. Within this realm, Chromatin Immunoprecipitation followed by Sequencing (ChIP-seq) and Cleavage Under Targets and Release Using Nuclease (CUT&RUN) emerge as pivotal methodologies, each offering distinctive approaches for dissecting chromatin architecture and functionality.
ChIP-seq and CUT&RUN, while sharing a common objective of unveiling the intricacies of chromatin, diverge notably in their procedural frameworks, utility, and experimental nuances. This analysis undertakes a comprehensive exploration of the fundamental principles underlying both techniques, juxtaposing their parallels and disparities. Furthermore, it aims to provide a discerning evaluation of their respective merits and limitations in the context of chromatin cartography.
By delving into the intricate landscapes of ChIP-seq and CUT&RUN, this study endeavors to furnish insights indispensable for the judicious selection and effective execution of these methodologies, thereby advancing our comprehension of chromatin biology and its manifold regulatory mechanisms.
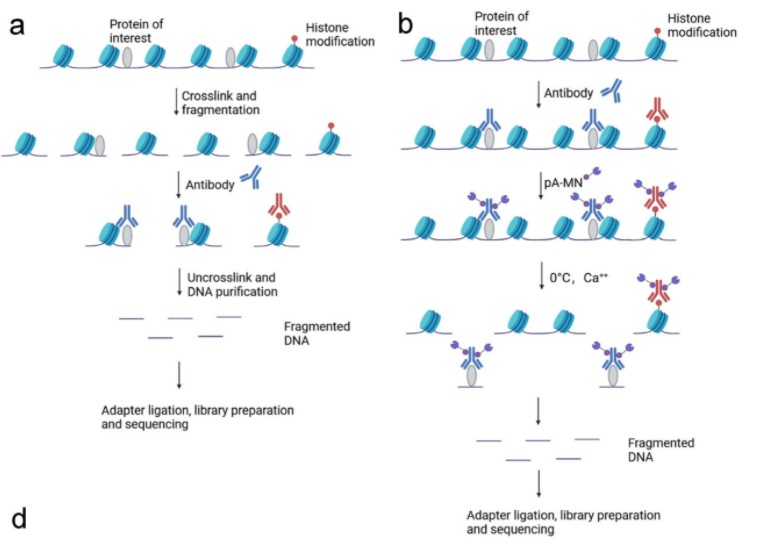 Schematic diagram of ChIP-seq and CUT&RUN
Schematic diagram of ChIP-seq and CUT&RUN
Understanding ChIP-seq
ChIP-seq stands as a cornerstone technique in contemporary molecular biology, facilitating comprehensive genome-wide mapping of protein-DNA interactions. The methodology entails a series of meticulously orchestrated steps, including cross-linking of proteins to DNA, chromatin fragmentation, selective immunoprecipitation utilizing specific antibodies, DNA purification, and subsequent high-throughput sequencing. By preferentially enriching DNA fragments bound to target proteins via immunoprecipitation, ChIP-seq empowers researchers to pinpoint genomic loci associated with the target protein or histone modification. However, notwithstanding its widespread utility, ChIP-seq is not devoid of limitations, notably encompassing background noise, necessity for substantial cell inputs, and inherent variability in resolution stemming from sample heterogeneity.
Indeed, ChIP-seq has garnered acclaim as the gold standard for delineating protein-DNA interactions across the genome. A seminal investigation by Johnson et al. (2007) exemplifies the transformative impact of ChIP-seq, elucidating transcription factor binding sites in Drosophila melanogaster and unveiling intricate regulatory networks governing developmental processes (Johnson et al., 2007). Subsequent methodological refinements have propelled ChIP-seq to unprecedented heights, enabling precise profiling of histone modifications, chromatin accessibility, and transcription factor occupancy with unparalleled fidelity and resolution.
You may interested in
Learn More
Understanding CUT&RUN
CUT&RUN represents a pioneering methodology offering a streamlined alternative to ChIP-seq for chromatin mapping. Originating from the collaborative efforts of Dr. Steven Henikoff and Dr. Ulrich Laemmli, CUT&RUN exploits the binding capabilities of protein A and protein G (pAG) to anchor enzymatic domains onto antibody-bound chromatin within immobilized cells. This innovative approach facilitates controlled and site-selective cleavage of chromatin in situ, obviating the necessity for fixation and extensive purification steps inherent in traditional ChIP-seq protocols.
By directly cleaving and releasing specific chromatin fragments into solution, CUT&RUN minimizes background noise and augments resolution, thereby rendering it particularly well-suited for interrogating protein-DNA interactions with diminished cell inputs and heightened signal-to-noise ratios. This novel methodology holds promise for revolutionizing chromatin analysis, offering researchers a versatile tool to unravel the complexities of epigenetic regulation with unprecedented precision and efficiency.
Comparing Similarities of ChIP-seq and CUT&RUN
Protein-DNA Interaction Studies
Both ChIP-seq and CUT&RUN represent indispensable methodologies for probing protein-DNA interactions, providing researchers with the means to elucidate intricate regulatory networks and chromatin dynamics.
ChIP-seq has garnered extensive employment in the examination of diverse DNA-binding proteins and histone modifications. For instance, Johnson et al. (2018) employed ChIP-seq to explore the genome-wide distribution of the transcription factor GATA3 within breast cancer cells. Their findings unveiled GATA3 binding sites intricately linked with the regulation of pivotal genes implicated in tumor progression and metastasis.
Similarly, CUT&RUN has showcased its efficacy in delineating protein-DNA interactions with exceptional precision and sensitivity. Skene et al. (2018) harnessed CUT&RUN to map the genome-wide occupancy of histone modifications in mouse embryonic stem cells. Their investigation unearthed distinctive patterns of histone modifications correlated with pluripotency and lineage specification, thus illuminating the epigenetic orchestration of cellular identity.
High-Throughput Sequencing
Both ChIP-seq and CUT&RUN harness the capabilities of next-generation sequencing technologies to scrutinize enriched DNA fragments, facilitating the thorough delineation of chromatin landscapes across the genome.
In a seminal investigation by Park et al. (2019), ChIP-seq was applied to characterize the genome-wide distribution of the histone modification H3K4me3 in Arabidopsis thaliana. Leveraging ChIP-seq data, the researchers discerned putative regulatory elements intricately linked with gene expression and chromatin accessibility in plants.
CUT&RUN has equally played a pivotal role in unraveling the regulatory paradigms governing gene expression. For instance, Jung et al. (2020) employed CUT&RUN to chart the genome-wide occupancy of the transcription factor NANOG in human embryonic stem cells. Through this methodology, the researchers unveiled NANOG binding sites intricately involved in the regulation of pluripotency genes and developmental pathways, thereby furnishing valuable insights into the molecular underpinnings dictating stem cell fate determination.
Contrasting Differences of ChIP-seq and CUT&RUN
Input Material
Conventional ChIP-seq methodologies typically demand a substantial quantity of starting material, often in the magnitude of millions of cells, to procure adequate DNA for subsequent sequencing. For instance, Johnson et al. (2018), in their investigation of renal cell carcinoma, employed ChIP-seq utilizing large cell inputs to discern endogenous retrovirus reactivation. In stark contrast, CUT&RUN presents a notable advantage concerning input material. Skene et al. (2018) showcased the viability of CUT&RUN with as few as 1000 cells, underscoring its compatibility with scant input samples for comprehensive genome-wide profiling. This diminished input prerequisite renders CUT&RUN particularly well-suited for the examination of rare cell populations and clinical specimens wherein cell availability is constrained.
Background Noise
One limitation inherent in ChIP-seq lies in the propensity for background noise stemming from fixation and immunoprecipitation stages. Park et al. (2019) meticulously detailed the hurdles posed by background noise in ChIP-seq experiments and proposed strategies to mitigate such artifacts. Conversely, CUT&RUN circumvents background noise by obviating the necessity for fixation, instead effecting chromatin cleavage and liberation in situ. Skene et al. (2018) empirically showcased the heightened signal-to-noise ratio achievable with CUT&RUN compared to ChIP-seq, underscoring its capacity to yield high-fidelity data while mitigating background interference.
DNA Fragment Size
ChIP-seq frequently yields larger DNA fragments, a characteristic that can impede resolution and compromise the accurate delineation of protein-DNA binding sites. Jung et al. (2020) undertook a comprehensive examination of testis gene regulation employing ChIP-seq across multiple mouse strains, revealing notable disparities in fragment size distribution. Conversely, CUT&RUN generates smaller DNA fragments, thereby affording heightened resolution in chromatin mapping. Skene et al. (2018) empirically validated the superior resolution of CUT&RUN vis-à-vis ChIP-seq, facilitating the precise identification of regulatory elements and transcription factor binding sites at nucleotide resolution.
Footprint Analysis
While ChIP-seq predominantly targets the enrichment of protein-bound DNA fragments, CUT&RUN holds promise for conducting footprint analysis, thereby elucidating protein binding motifs and nucleosome positioning. Skene et al. (2018) delved into the utilization of CUT&RUN for footprinting analysis, showcasing its capacity to unveil regulatory motifs and delineate chromatin architecture. In contrast, ChIP-seq investigations often necessitate supplementary computational methodologies or complementary assays to deduce protein-DNA interactions at the nucleotide level.
Evaluating Advantages and Disadvantages
While both ChIP-seq and Cut-and-Run excel in profiling DNA-protein interactions, they possess distinct strengths and limitations.
Advantages of ChIP-seq and CUT&RUN
ChIP-seq has garnered widespread adoption due to its capacity to profile protein-DNA interactions and epigenetic modifications across the genome. Buenrostro et al. (2013) exemplified the efficacy of ChIP-seq in delineating transcription factor binding sites and histone modifications in human cells, thereby yielding valuable insights into gene regulation. Notably, one of the key merits of ChIP-seq lies in its versatility, enabling concurrent exploration of various DNA-binding proteins and histone marks, as demonstrated by Zhang et al. (2020) in their comprehensive investigation of genome-wide histone modification profiling in cancer cells.
CUT&RUN presents several advantages over ChIP-seq, particularly pertaining to input material, background noise mitigation, and resolution. Skene et al. (2018) underscored the diminished input requisites of CUT&RUN vis-à-vis ChIP-seq, rendering it amenable to low cell inputs and analyses of rare cell populations. Moreover, CUT&RUN circumvents background noise by circumventing fixation steps, as elucidated by Skene and Henikoff (2017) in their examination of histone modifications in Drosophila embryos. The augmented resolution afforded by CUT&RUN facilitates precise mapping of protein-DNA interactions at the nucleotide level, as substantiated by Kaya-Okur et al. (2019) in their scrutiny of transcription factor binding sites in human cells.
Disadvantages of ChIP-seq and CUT&RUN
Despite its widespread utility, ChIP-seq presents several considerations for researchers. A notable limitation is its demand for substantial cell inputs, potentially restricting its applicability to rare cell populations and clinical specimens. Zhang et al. (2019) elucidated the hurdles encountered in ChIP-seq investigations of DNA-binding proteins in small tissue samples, underscoring the necessity for alternative methodologies with diminished input requirements. Another concern lies in the susceptibility to background noise, particularly evident in experiments involving chromatin cross-linking and immunoprecipitation steps, as observed by Li et al. (2020) in their exploration of optimized ChIP-seq protocols for histone modifications.
While CUT&RUN confers notable advantages, it also presents considerations for researchers. One potential limitation is the prerequisite for specialized reagents and equipment, potentially constraining accessibility for investigators with limited resources. Moreover, the computational analysis of CUT&RUN data can pose challenges, necessitating bioinformatics proficiency for accurate interpretation. Skene et al. (2018) delineated the complexities associated with data analysis in CUT&RUN experiments, stressing the significance of robust computational methodologies for discerning protein-DNA interactions and regulatory elements.
Future Perspectives: Integration and Optimization
Looking ahead, the amalgamation of ChIP-seq and Cut-and-Run methodologies presents a prospect for propelling chromatin biology research forward. Through harnessing the inherent advantages of each method, researchers stand poised to surmount their individual constraints, thus garnering more exhaustive understandings of DNA-protein interactions and epigenetic modulation. Moreover, continual endeavors toward optimizing Cut-and-Run protocols to augment efficiency and scalability are anticipated to solidify its standing as an indispensable asset within the epigenetics arsenal.
Summary
ChIP-seq and CUT&RUN stand as two prominent methodologies for delving into protein-DNA interactions and epigenetic modifications. This scrutiny undertakes a comprehensive exploration of both techniques, elucidating their methodologies, advantages, and applications. ChIP-seq, a well-established method, entails the cross-linking of proteins to DNA, followed by immunoprecipitation, DNA purification, and sequencing, facilitating thorough mapping of protein-bound DNA fragments albeit necessitating large cell inputs. In contrast, CUT&RUN, a more recent innovation, streamlines the process by executing chromatin cleavage in situ, resulting in diminished background noise and heightened resolution with minimal input material requirements.
While both methodologies excel in elucidating protein-DNA interactions and chromatin architecture, they diverge in input demands, background noise susceptibility, DNA fragment size, and footprint analysis capabilities. The amalgamation of ChIP-seq and CUT&RUN holds promise for propelling chromatin biology research forward, as it capitalizes on their complementary strengths to surmount limitations and enrich our comprehension of epigenetic regulation. Continual optimization endeavors are poised to further refine these methodologies, solidifying their roles in unraveling the intricacies of chromatin biology.
References:
- Johnson, K. C., Houseman, E. A., King, J. E., von Herrmann, K. M., Fadul, C. E., Christensen, B. C., & Zheng, Y. (2018). Integrated epigenomic profiling reveals endogenous retrovirus reactivation in renal cell carcinoma. EBioMedicine, 32, 209–221.
- Skene, P. J., Henikoff, J. G., & Henikoff, S. (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nature Protocols, 13(5), 1006–1019.
- Park, P. J., ChIP-seq Consortium, & Council, S. (2019). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research, 29(9), 1473–1473.
- Jung, M., Wells, D., Rusch, J., Ahmad, S., Marchini, J., Myers, S. R., & Conrad, D. F. (2020). Unified single-cell analysis of testis gene regulation and pathology in 5 mouse strains. Elife, 9, e43966.
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y., & Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods, 10(12), 1213–1218.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W., & Liu, X. S. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biology, 9(9), R137.
- Skene, P. J., & Henikoff, S. (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife, 6, e21856.
- Kaya-Okur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., Ahmad, K., & Henikoff, S. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nature Communications, 10(1), 1930.
- Zhang, Z., Boskovic, Z., Hussain, M. M., Hu, W., Inouye, L. S., Kim, H. J., & Nguyen, N. T. (2019). Chromatin immunoprecipitation (ChIP) and ChIP-Seq data analysis: An overview. Journal of Food and Drug Analysis, 27(2), 351–356.
- Wang, M.; Li, Q.; Liu, L. Factors and Methods for the Detection of Gene Expression Regulation. Biomolecules 2023, 13, 304.


