
 Sample Submission Guidelines
Sample Submission Guidelines
What Is Cappable-Seq? Principle, Workflow, Advantages, Applications
Accurately determining where transcription begins is essential for understanding bacterial gene regulation and promoter function, yet conventional RNA-seq cannot distinguish true initiation sites from processed RNA. Cappable-Seq directly addresses this limitation by selectively capturing 5′-triphosphate primary transcripts, enabling precise and quantitative transcription start site (TSS) mapping at single-nucleotide resolution. This article outlines the underlying principles, experimental workflow, and data analysis strategies of Cappable-Seq, and compares it with related TSS-profiling methods to illustrate how this approach is key to decoding bacterial regulatory logic, with broad implications from fundamental research to biotechnology.
1. Introduction
Accurate characterization of transcription initiation is essential for dissecting bacterial gene regulation, promoter architecture, and cellular responses to environmental changes. Although conventional RNA sequencing offers a snapshot of steady-state RNA levels, it cannot discriminate primary transcripts that retain the native 5′-triphosphate (5′-PPP) group from RNAs that have undergone processing or degradation.
Cappable-Seq addresses this challenge through a highly selective 5′-end enrichment strategy designed for single-nucleotide TSS profiling. By recognizing the distinctive 5′-PPP signature on nascent RNA, this approach selectively enriches primary transcripts with remarkable specificity. As a result, Cappable-Seq has emerged as a crucial tool in microbial transcriptomics and is now increasingly applied across biotechnology, synthetic biology, and therapeutic development. For organizations engaged in microbial strain engineering or fermentation process development, this method offers crucial visibility into promoter activity and regulatory dynamics that standard RNA sequencing cannot reveal.
Cappable-Seq is widely used for high-resolution transcription start site (TSS) profiling across bacterial genomes, including applications in bacterial TSS profiling, 5′-triphosphate RNA enrichment, and primary transcript enrichment technology.
 Figure 1. mRNA caps in eukaryotes
Figure 1. mRNA caps in eukaryotes
2. Biochemical Principle of Cappable-Seq
Cappable-Seq is built upon a precise enzymatic reaction that relies on the specificity of the Vaccinia Capping Enzyme (VCE). VCE inherently recognizes RNA molecules bearing a 5′-triphosphate and catalyzes the transfer of a guanosine cap analog to their 5′ termini. In this workflow, the reaction incorporates a modified cap analog bearing an abiotin tag, enabling selective isolation of authentic 5′-PPP RNAs. Through this design, the method enables accurate delineation of transcription initiation sites and supports applications that depend on targeted enrichment of 5′-triphosphate RNA or sequencing of primary transcripts.
The underlying mechanism unfolds through three distinct enzymatic steps:
- RNA Triphosphatase Activity– VCE removes the γ-phosphate from 5′-PPP
- Guanylyltransferase Activity– VCE adds a GMP moiety to the 5′-diphosphate
- (Modified) Cap Transfer– A biotinylated GTP analog is incorporated instead of natural methylation
Only genuine primary transcripts undergo complete labeling, ensuring high specificity for transcription initiation sites. Biotin-tagged RNAs are then captured using streptavidin-coated beads, effectively separating them from processed RNA and degradation fragments. The enriched RNA obtained through this procedure provides a highly suitable input for subsequent cDNA synthesis and library preparation steps.
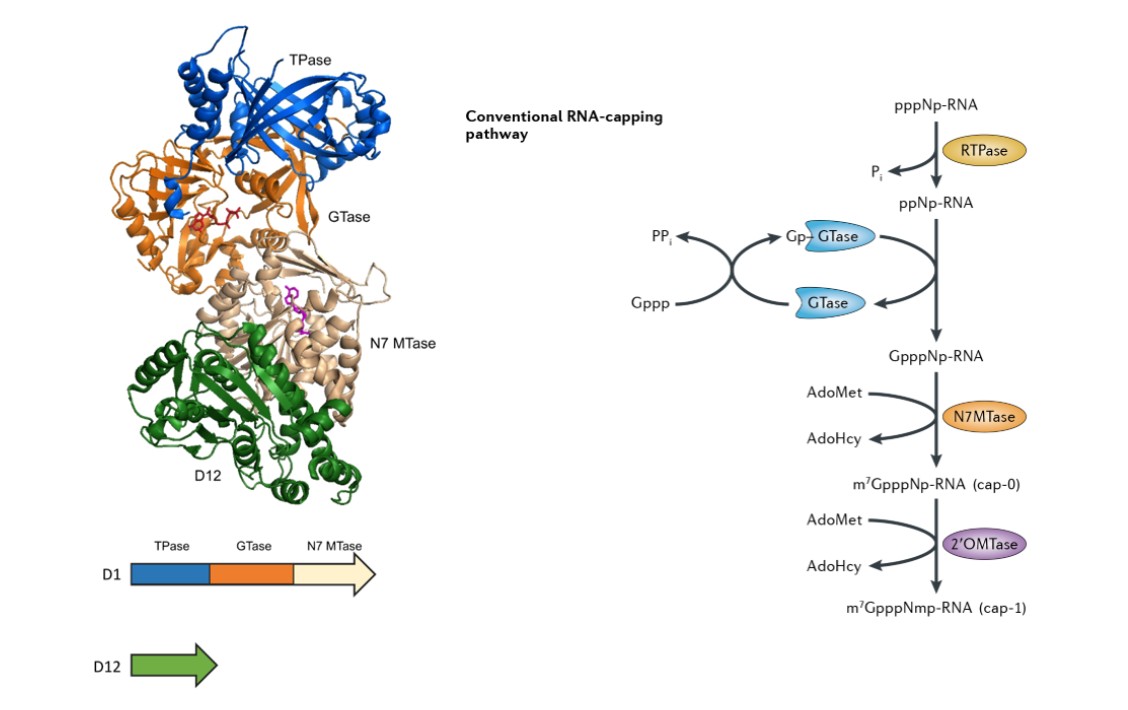 Figure 2. Structure and Reaction Mechanism of the Vaccinia Virus Capping Enzyme
Figure 2. Structure and Reaction Mechanism of the Vaccinia Virus Capping Enzyme
3. Experimental Workflow
A standard Cappable-Seq protocol comprises these key steps. This enrichment approach has become a standard component in advanced applications aimed at profiling transcription start sites across bacterial transcriptomes:
3.1 RNA Preparation
High-quality total RNA is first obtained through phenol–chloroform extraction or column-based purification. Next, thorough DNase treatment removes any residual genomic DNA. It is then essential to maintain strong RNA integrity (RIN > 8.0) to prevent overrepresentation of degraded RNA fragments. Throughout the process, pay special attention to RNA stability to preserve the 5′-PPP groups.
To limit oxidative degradation and preserve the native 5′-triphosphate structure of primary transcripts, it is commonly recommended to include approximately 1% β-mercaptoethanol in the lysis buffer during bacterial RNA extraction. Extended DNase digestion is often applied to further reduce residual genomic DNA, which could otherwise introduce artifactual transcription start site signals during downstream analyses. With these precautions in place, the workflow moves to the selective capping reaction.
3.2 Selective Capping Reaction
Purified RNA is incubated with VCE and biotinylated cap analog under optimized buffer conditions. Following incubation, primary transcripts are selectively tagged, leaving processed RNAs unlabeled. Throughout this step, conditions are carefully controlled to maximize labeling efficiency while minimizing nonspecific labeling.
For RNA samples with substantial secondary structure, a brief heat-denaturation step (e.g., 70°C for ~2 minutes) prior to VCE addition is often recommended in methodological literature to improve enzyme accessibility to 5′-PPP termini and enhance overall capping efficiency. Following capping optimization, the workflow proceeds to capture the biotinylated RNAs.
3.3 Affinity Capture
Biotinylated RNAs are captured using streptavidin-coated magnetic beads, followed by a series of stringent wash steps. High-salt wash conditions (0.5–1.0 M NaCl) are commonly used to minimize non-specific RNA–bead interactions and to enhance enrichment purity. Allowing a brief settling period during the final wash can further help remove RNA fragments that remain only weakly associated. Once capture and washing are complete, the workflow proceeds to library construction.
3.4 Library Construction
Specific adapters are ligated to the 3′ end of the enriched RNA, after which reverse transcription is carried out using specialized enzymes. Employing reverse transcriptases that lack terminal transferase activity is essential to maintain the precise 5′ end information during cDNA synthesis. Prior to sequencing, library quality is evaluated to ensure that the final datasets meet required performance standards. After confirming quality, sequencing can begin.
3.5 Sequencing
Final libraries are sequenced on Illumina platforms, providing single-nucleotide resolution of transcription start sites. In most bacterial studies, generating approximately 10–20 million reads per sample offers sufficiently comprehensive coverage, though the required depth can vary with genome complexity and the goals of the experiment.
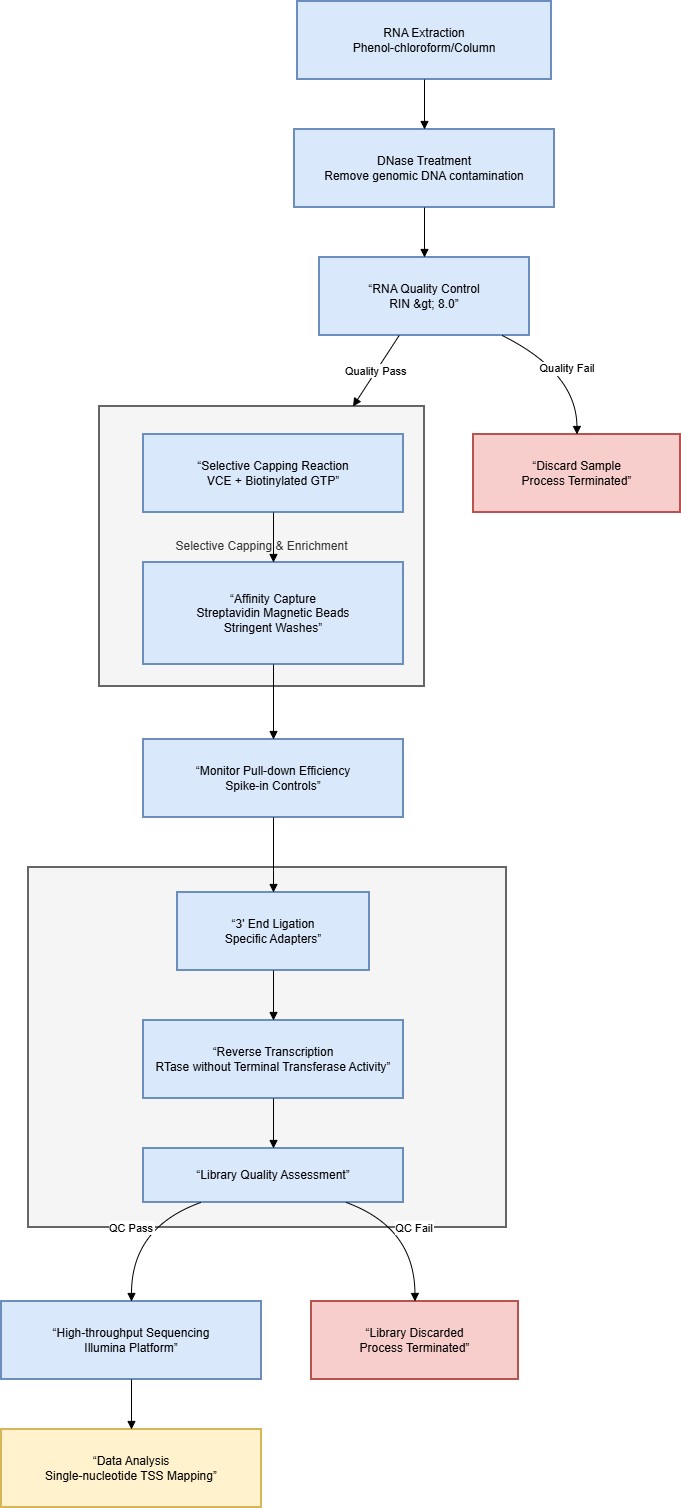 Figure 3. Cappable-Seq library preparation workflow
Figure 3. Cappable-Seq library preparation workflow
4. Data Analysis Pipeline
Robust bioinformatics processing is essential for accurate TSS identification and interpretation:
4.1 Quality Control
FastQC assesses overall read quality, GC content, adapter contamination, and sequence complexity. This preliminary evaluation enables early detection of potential issues in the analysis pipeline and ensures that the dataset meets quality requirements for subsequent processing.
4.2 Read Processing
Cutadapt or Trimmomatic removes adapters, low-quality bases, and short fragments. Parameters are optimized based on library preparation details and sequencing quality metrics to maximize retention of meaningful sequence data while eliminating technical artifacts.
4.3 Genome Alignment
Bowtie2 efficiently maps reads to bacterial reference genomes, and alignment rates above 90% generally indicate effective enrichment. For more complex microbial communities or eukaryotic samples, tools such as STAR or other splice-aware aligners can be used to accommodate alternative splicing events.
4.4 TSS Identification
The first nucleotide of each mapped read marks a potential TSS location. Genomic regions with multiple 5′ ends form clear peaks, interpreted as candidate transcription start sites. Statistical approaches are then used to differentiate genuine TSSs from the background signal.
4.5 TSS Classification
Specialized algorithms categorize TSSs based on their genomic context and expression levels:
- Primary TSSs: Dominant transcription initiation sites for genes
- Secondary TSSs: Alternative start sites with lower activity
- Internal TSSs: Start within coding sequences, often indicating regulatory complexity
- Antisense TSSs: Initiation on opposite strands, suggesting regulatory RNA production
4.6 Genomic Context Analysis
Identified TSSs are annotated relative to known genomic features, including promoters, operons, coding sequences, and regulatory elements. This contextual information helps interpret the biological significance of identified start sites.
4.7 Visualization and Interpretation
IGV, along with custom R or Python scripts, supports visual inspection of TSS peaks and their surrounding genomic context. More advanced visualization strategies can integrate additional data layers, providing a broader, more detailed view of transcriptional regulation.
5. Technical Comparisons
Cappable-Seq demonstrates distinct advantages over alternative TSS mapping approaches due to its unique positive selection mechanism and high specificity for 5′-triphosphate RNA. The following comparison highlights key technical features and performance metrics relative to other commonly used methods, providing a clear rationale for its adoption in various experimental contexts.
Table 1: Technology Comparison
| Feature | Cappable-Seq | dRNA-Seq | 5'-RACE | SMRT-Cappable |
|---|---|---|---|---|
| Principle | Positive selection | Negative selection | PCR amplification | Long-read integration |
| Specificity | Very High | High | High | Very High |
| Throughput | Genome-wide, High | Genome-wide, High | Gene-specific, Low | Genome-wide, Medium |
| Resolution | Single-base | Single-base | Single-base | Single-base + Full-length |
| Advantage | Low background, quantitative | Established protocol | Equipment simple | Structural context |
| Limitation | Biotin handling, optimization required | Enzyme bias, incomplete digestion | Low throughput, not discovery-based | Cost, computational complexity |
5.1 Cappable-Seq vs dRNA-Seq
While both technologies target primary transcripts, their enrichment strategies differ fundamentally. dRNA-Seq employs Terminator exonuclease (TEX) to degrade processed RNA (negative selection), whereas Cappable-Seq directly labels and captures 5′-PPP RNA (positive selection). This distinction gives Cappable-Seq superior signal-to-noise characteristics, as TEX digestion efficiency can vary with RNA secondary structure and may not completely remove all processed fragments. Additionally, Cappable-Seq's positive selection approach provides more consistent results across different RNA species and experimental conditions.
5.2 Cappable-Seq vs 5′-RACE
5′-RACE provides precise TSS validation for individual genes but lacks the scalability required for genome-wide studies. The method is labor-intensive and not suitable for discovery-based approaches. In contrast, Cappable-Seq enables comprehensive TSS discovery across entire genomes in a single experiment, supporting comparative analysis across multiple conditions, time points, and genetic backgrounds. The sequencing-based nature of Cappable-Seq also provides quantitative information about TSS usage levels, enabling more sophisticated regulatory analyses.
5.3 Cappable-Seq vs Long-Read Integration
SMRT-Cappable-Seq combines Cappable-Seq specificity with PacBio long-read sequencing, simultaneously providing TSS mapping and full transcript structure information. This integrated approach is particularly valuable for studying complex transcriptional architectures, alternative splicing, and operon organization. However, the significantly higher costs, lower throughput, and substantial computational requirements make short-read Cappable-Seq more practical for most large-scale screening applications and routine profiling studies.
Cappable-Seq offers the optimal balance of specificity, resolution, quantitative performance, and practical feasibility for comprehensive bacterial TSS profiling. The choice between technologies should consider specific research objectives, available resources, and the required balance between discovery depth and analytical resolution.
6. Research Applications
Cappable-Seq enables diverse transcriptomic investigations across multiple research domains:
6.1 Comprehensive Promoter Identification and Characterization
The precise mapping of transcription start sites enables accurate promoter identification and regulatory motif discovery. In some studies, Cappable-Seq defined promoter architecture across metabolic genes, revealing not only canonical -10 and -35 boxes but also identifying alternative sigma factor binding sites. The technology's single-nucleotide resolution allows researchers to identify overlapping promoters and assess their relative strengths under different physiological conditions. This detailed promoter mapping is particularly valuable for synthetic biology applications, where precise promoter engineering requires accurate knowledge of transcription initiation points and regulatory contexts.
6.2 Operon Structure Delineation and Transcriptional Organization
By defining precise transcription start sites, Cappable-Seq enables accurate determination of operon boundaries and reveals complex regulatory architectures. Research in Escherichia coli demonstrated that previously annotated operons often contain internal promoters that allow differential expression of genes within polycistronic units. This finding has significant implications for understanding bacterial physiology, as it reveals additional layers of transcriptional control beyond classical operon models. Comparative Cappable-Seq analyses across growth conditions have uncovered condition-specific operon structures, providing insights into how bacteria dynamically reorganize their transcriptional units in response to environmental changes.
6.3 Discovery and Characterization of Regulatory RNAs
The technology's high sensitivity makes it exceptionally powerful for identifying small regulatory RNAs (sRNAs) and antisense RNAs, which are often expressed at low levels and originate from intergenic regions. A comprehensive analysis of the human gut microbiome using Cappable-Seq uncovered hundreds of previously unannotated sRNAs, many showing conservation across bacterial species and potential involvement in host-microbe interactions. Beyond discovery, Cappable-Seq enables functional characterization by precisely mapping start sites and identifying potential regulatory targets through genomic context analysis.
6.4 Bacterial Stress Response and Adaptation Mechanisms
Cappable-Seq provides unique insights into how bacteria reprogram their transcriptomes in response to environmental challenges. Studies characterized complex transcriptional remodeling under antibiotic stress, identifying novel stress-responsive promoters and non-coding RNAs contributing to survival mechanisms. The method's quantitative nature enables tracking dynamic TSS usage changes over time, revealing how bacterial cells sequentially activate different regulatory programs during adaptation. This application extends to industrial contexts, where understanding microbial responses to process-related stresses can inform bioprocess optimization.
6.5 Genome Annotation Enhancement and Comparative Genomics
For newly sequenced microbial genomes, Cappable-Seq provides experimental evidence for gene boundaries and reveals transcriptionally active regions missed by computational prediction. The pioneering application in Helicobacter pylori led to major annotation revisions, accurately defining translation start sites and discovering extensive non-coding transcription. In comparative genomics, Cappable-Seq datasets from related strains reveal conservation and divergence in regulatory strategies, providing insights into evolutionary adaptations and niche specialization.
6.6 Metabolic Engineering and Synthetic Biology Applications
In industrial biotechnology, Cappable-Seq has emerged as a powerful tool for characterizing and engineering microbial production strains. By providing comprehensive transcription initiation maps, the technology enables rational design of synthetic promoters and identification of native regulatory elements for metabolic engineering. Applications include cyanobacterial strain optimization for biofuel production, where Cappable-Seq identified strong native promoters for heterologous pathway expression. The technology also helps identify and eliminate cryptic promoters causing metabolic imbalances in engineered strains.
6.7 Host-Pathogen Interactions and Virulence Regulation
Cappable-Seq provides crucial insights into how bacterial pathogens coordinate virulence gene expression during infection. Studies in Salmonella typhimurium and Listeria monocytogenes mapped complex regulatory networks controlling virulence factor expression in response to host environmental cues. The high resolution has been particularly valuable for identifying small regulatory RNAs that fine-tune virulence gene expression, revealing potential targets for anti-virulence therapies. Comparative TSS mapping between laboratory conditions and infection models identifies infection-specific promoters activated during host-pathogen interactions.
6.8 Antibiotic Resistance Mechanisms and Persister Cell Formation
The technology advances our understanding of how bacteria regulate the expression of antibiotic resistance genes and how transcriptional heterogeneity contributes to persister cell formation. Studies of Pseudomonas aeruginosa and Staphylococcus aureus revealed complex promoter architectures that control multidrug efflux pumps and other resistance determinants. By enabling single-nucleotide-resolution TSS mapping in bacterial populations, the technology helps identify subpopulations with distinct transcriptional programs that contribute to antibiotic tolerance, providing insights into mechanisms that could be targeted to restore antibiotic efficacy.
7. Conclusion and Outlook
Cappable-Seq has revolutionized prokaryotic transcriptomics by providing an unparalleled combination of specificity, resolution, and quantitative performance. By directly targeting the 5′-triphosphate signature of nascent RNA, it reveals genuine transcription initiation events that are typically obscured in standard RNA-seq data, delivering unprecedented insights into bacterial regulatory architecture.
Looking forward, the integration of Cappable-Seq with long-read sequencing technologies promises to enhance its capabilities, enabling simultaneous TSS mapping and full-length transcript analysis. The development of single-cell adaptations will further advance our understanding of transcriptional heterogeneity in bacterial populations.
As protocols become more streamlined and accessible, applications will continue to expand into clinical pathogen profiling and industrial strain optimization. Cappable-Seq remains positioned as a cornerstone technology for deciphering bacterial regulatory logic, supporting both fundamental research and biotechnological innovation across multiple domains.
Next steps you can take now:
- Contact us to discuss your bacterial strain and project requirements
- Request a customized quote for Cappable-Seq based on your sequencing needs
- Explore integrating Cappable-Seq with other omics approaches for comprehensive analysis
Our experts provide end-to-end solutions from experimental design to data interpretation, supporting your bacterial transcriptional regulation research.
8. Frequently Asked Questions (FAQs)
Q1: What is the main advantage of Cappable-Seq over standard RNA-Seq for TSS mapping?
A1: Unlike standard RNA-Seq, Cappable-Seq specifically enriches for primary transcripts bearing 5′-triphosphate ends, allowing precise mapping of TSSs and distinguishing them from processed RNA fragments.
Q2: Can Cappable-Seq be applied to eukaryotic organisms?
A2: The application of Cappable-Seq in eukaryotes is limited by fundamental biological differences. The hallmark 5′-triphosphate terminus of bacterial primary transcripts is transient in most eukaryotic mRNAs due to immediate co-transcriptional processing. While this limits the direct utility of the standard protocol, the general principle of selecting for native 5′ ends has been explored in limited eukaryotic contexts, often integrated with long-read transcriptome analyses. However, it is important to note that these approaches are still in developmental stages and have not reached the level of establishment seen in prokaryotic research.
Q3: How does Cappable-Seq compare to dRNA-Seq in terms of specificity?
A3: Cappable-Seq employs a positive selection strategy (labeling and capturing 5′-PPP RNA), which generally provides higher specificity and a better signal-to-noise ratio compared to the negative selection approach (degrading processed RNA) used in dRNA-Seq.
Q4: What are the key experimental considerations for a successful Cappable-Seq experiment?
A4: Critical factors include: 1) high RNA integrity (RIN > 8.0); 2) efficient DNase treatment to remove genomic DNA; 3) use of β-mercaptoethanol during extraction to preserve 5′-PPP groups; and 4) optimization of the capping reaction conditions, including potential heat-denaturation for structured RNAs.
References:
- Bischler, T., Tan, H. S., Nieselt, K., & Sharma, C. M. (2015). Differential RNA-seq (dRNA-seq) for annotation of transcriptional start sites and small RNAs in Helicobacter pylori. Methods. https://doi.org/10.1016/j.ymeth.2015.06.012
- Yan, B., Boitano, M., Clark, T. A., & Ettwiller, L. (2018). SMRT-Cappable-seq reveals complex operon variants in bacteria. Nature communications. https://doi.org/10.1038/s41467-018-05997-6
- Cortes, T., Schubert, O. T., Rose, G., Arnvig, K. B., Comas, I., & Aebersold, R. (2013). Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in Mycobacterium tuberculosis. Cell Reports. https://doi.org/10.1016/j.celrep.2013.10.031
- Ettwiller, L., Buswell, J., Yigit, E., & Schildkraut, I. (2016). A novel enrichment strategy reveals unprecedented number of transcription start sites at single-base resolution in a model prokaryote and the gut microbiome. Microbial Genomics. https://doi.org/10.1186/s12864-016-2539-z
- Kyrieleis, O. J., Chang, J., de la Peña, M., Shuman, S., & Cusack, S. (2014). Crystal structure of vaccinia virus mRNA capping enzyme provides insights into the mechanism and evolution of the capping apparatus. Structure. https://doi.org/10.1016/j.str.2013.12.014
- Ramanathan, A., Robb, G. B., & Chan, S. H. (2016). mRNA capping: Biological functions and applications. Nucleic Acids Research. https://doi.org/10.1093/nar/gkw551
- Sessegolo, C., Cruaud, C., Da Silva, C., et al. (2019).Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Scientific Reports. https://doi.org/10.1038/s41598-019-51470-9
- Movahedzadeh, F., Gonzalez-Y-Merchand, J. A., & Cox, R. A. (2001). Transcription start-site mapping. Totowa, NJ: Humana Press. https://doi.org/10.1385/1-59259-147-7:105
- Sharma, C. M., Hoffmann, S., Darfeuille, F., et al. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature. https://doi.org/10.1038/nature08756
- Thomason, M. K., Bischler, T., Eisenbart, S. K., Förstner, K. U., Zhang, A., & Vogel, J. (2015). Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. Journal of Bacteriology. https://doi.org/10.1128/JB.02096-14
- Wang, Z., Gerstein, M., & Snyder, M. (2009). RNA-Seq: A revolutionary tool for transcriptomics. Nature Reviews Genetics. https://doi.org/10.1038/nrg2484
- Agustinho, D. P., Fu, Y., Menon, V. K., Metcalf, G. A., Treangen, T. J., & Sedlazeck, F. J. (2024). Unveiling microbial diversity: harnessing long-read sequencing technology. Nature methods. https://doi.org/10.1038/s41592-024-02262-1


