
 Sample Submission Guidelines
Sample Submission Guidelines
The Power of BSA: Advancing Trait Localization Research
BSA (Bulk segregant analysis), called hybrid grouping analysis, is a rapid and simple method for trait localization that has emerged in recent years. One of the prominent features of BSA is the pool-seq of the offspring of two extreme traits. However, BSA is not as simple as mixing sequencing. In practical applications, there are various methods to resolve the differences in population materials and experimental design.
What is BSA (Bulked Segregant Analysis)?
BSA (Bulked Segregant Analysis), initially pioneered by R.W. MICHELMORE in 1991, has emerged as a powerful methodology for swiftly pinpointing genes governing specific traits. The core principle revolves around selecting parents displaying substantial phenotypic disparities to establish a segregating or family population dedicated to the study of the target trait. Subsequently, a set number of individual plants exhibiting extreme phenotypes for the trait are chosen from the segregating population and combined to create two DNA "pools." The contrasting genetic variations within these pools indicate the potential candidate region where the gene or QTL of interest might be situated. Typically, the DNA of both parents is employed as a control during the comparison of differences between the two DNA pools, enabling a comprehensive analysis and accurate interpretation of experimental findings.
Please refer to Bulk Segregant Analysis Q&A for more information.
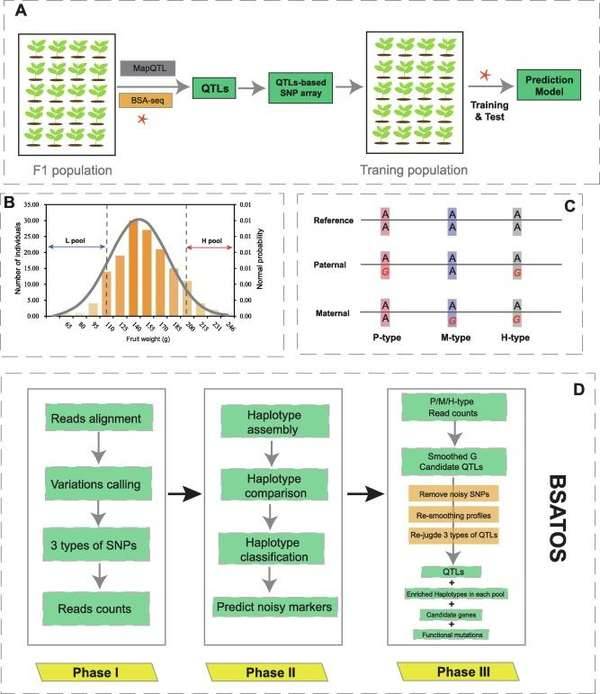 Bulked segregant analysis. (Shen et al., 2022)
Bulked segregant analysis. (Shen et al., 2022)
How to Do Bulked Segregant Analysis?
According to the origin of the target traits, BSA trait localization can be divided into two directions: natural traits (QTL-seq) and artificially mutated traits (mutant map, MutMap).
QTL-seq
QTL-seq employs a strategy where parents exhibiting extreme traits are carefully selected to establish a lineage. The offspring population, showcasing a normal distribution of the target traits, is segregated, and individuals displaying extreme traits at both ends of this distribution are chosen to form two equivalent sample pools. These pools are then subjected to sequencing, with subsequent analysis of SNP-index allowing for the identification of loci or regions associated with the target traits. Primarily, this method is employed in the identification of key effect genes underlying quantitative traits.
MutMap
MutMap entails the creation of a familial population through the backcrossing of mutant individuals with either the parents or distantly related inbred species. From this segregating offspring population, a subset of individuals exhibiting the mutant traits are selected to generate a gene pool. Simultaneously, a separate gene pool comprising a certain number of wild-type offspring is also formed to serve as a control for the mutant pool. The mixed samples from these pools are subsequently subjected to sequencing, and the obtained results are compared against the parental genome or the pool of offspring exhibiting wild traits. By examining the SNP-index, the loci or regions associated with the target traits are precisely identified. This method is particularly useful for detecting point mutations resulting from either chemical or physical mutagenesis and finds widespread application in the localization studies of quality traits.
Workflow of Bulked Segregant Analysis
- Phenotypic Selection: Discern a phenotypic characteristic of significance that exhibits distinct segregation between two discernible classes, such as the wild type and mutant.
- Crossbreeding and Generation of Segregating Population: Generate a population by crossing individuals exhibiting divergent phenotypes. For instance, in the case of studying a plant trait, mate a mutant individual possessing the desired phenotype with a wild-type individual.
- Phenotypic Screening and Preparation of Bulk Samples: Allow the offspring resulting from the crossbreeding to grow and mature. Conduct a phenotypic assessment of the progeny to identify individuals showcasing the desired phenotypic extremes. Specifically, select an equal number of individuals displaying the mutant phenotype and individuals exhibiting the wild-type phenotype.
- DNA Extraction: Extract genomic DNA from each chosen individual in both the mutant and wild-type pools separately. Employ an appropriate DNA extraction method to obtain DNA of superior quality.
- Pooling and Sequencing: Merge the extracted DNA from the mutant pool and the wild-type pool individually to produce two consolidated DNA samples. Employ high-throughput sequencing technologies, such as next-generation sequencing (NGS), to sequence the DNA of the mutant and wild-type samples.
- Data Analysis: Analyze the sequencing data to detect genetic variations, such as single nucleotide polymorphisms (SNPs), between the mutant and wild-type samples. Compute the SNP-index, which denotes the allele frequency of SNPs in each consolidated sample. This analysis will facilitate the identification of genomic regions associated with the targeted phenotype.
- Identification of Candidate Genes: Compare the SNP-index values across the entire genome to pinpoint regions or loci linked to the desired trait. Identify candidate genes within these regions that potentially contribute to the observed phenotype. Subsequently, validate these candidate genes through supplementary experiments and functional analyses.
Reference:
- Shen, Fei, et al. "A bulked segregant analysis tool for out-crossing species (BSATOS) and QTL-based genomics-assisted prediction of complex traits in apple." Journal of Advanced Research 42 (2022): 149-162.


