
 Sample Submission Guidelines
Sample Submission Guidelines
Introduction to Transcription Factors
Molecules important in controlling gene expression are transcription factors. Normally they are proteins, although they are composed of short, non-coding RNA as well. Transcription variables are also typically seen working in groups or complexes, creating multiple connections that allow varying degrees of control over transcription rates.
Transcription factors (TFs) attach to obtainable or "open" promoter and enhancer regions, which, by facilitating or hindering RNA polymerase binding, serve a crucial role in regulating gene expression. Various binding actions result in gene expression heterogeneity along with a cell population, which can give rise to specific cell identities. The supervisors of the cellular plant are transcription factors, which regulate everything from cellular individuality to reaction to external stimuli. Thousands of people are affected by mutations living within transcription factors due to their vital significance in analyzing the genome, causing a wide range of symptoms. In addition, many other illness-causing mutations, e.g. enhancers, which are abundant with TF binding sites, are discovered in regulatory sites. The characterization of TF binding profiles is thus critical for knowing gene regulatory systems and cell differentiation into separate subpopulations.
Techniques Used to Study Transcription Factor Binding Activity
Understanding of the two main roles of a transcription factor is required to analyze transcription factor interaction: binding to DNA and transcription reconfiguration. Transcription factors, a TF recognition motif, attach to specific DNA sequences. In order to determine and classify such recognition motifs, numerous methods have been used. A technique to assess transcription factor binding activity across the entire genome is to analyze chromatin accessibility assays such as deoxyribonuclease hypersensitive sites sequencing (DNase-seq), formaldehyde-assisted isolation of regulatory elements sequencing (FAIRE-seq), and transposase-accessible chromatin sequencing assay (ATAC-seq). Connections between protein and DNA can also be gauged genome-wide by chromatin immunoprecipitation preceded by sequencing (ChIP-seq). Multiple pieces of proof, unfortunately, imply that not all binding circumstances affect transcription.
Another approach to identifying individual protein-DNA binding areas is via DNA footprinting to deduce a large number of binding events. Small regions safeguarded from cleavage by the existence of a bound transcription factor are identified by dense mapping of DNase I cleavage locations. While a large collection of recently undescribed motifs guarded against cleavage was defined by initial footprinting research, implying many novel transcription factors, current studies suggest that these regions probably influence the DNase I enzyme’s sequence-based cleavage bias. Furthermore, it is now also evident that most TFs (80%) do not exhibit a quantifiable footprint, thus restricting the effectiveness of this method.
Some of the earliest research of TFs as regulators were according to expression data, because TFs modify transcription. Large compilations of expression information have been used to deduce gene regulatory networks for nearly two decades. Usually, these methods search for components, compilations of co-regulated genes across different circumstances. Specific TFs are linked to the gene module they control by the detection of nearby TF recognition motifs or co-regulated transcription factors. The techniques of the gene regulatory network have been involved in interpreting large-scale regulatory networks but are innately restricted by the reality that they rely on information for steady-state expression. Assays of steady-state expression (microarray or RNA-seq) not only represent transcription, but also the processing, maturation, and stability of RNA. Therefore, they are an indirect reading of the impact of disturbances on transcription variables. In relation, without an unrealistic number of replicates, they are generally unable to reliably detect small changes at short time points. Nascent transcription assays effectively describe RNA associated with cellular polymerases involved (GRO-seq and PRO-seq). As a result, nascent assays are a direct reading of transcription modifications caused by disturbances.
The ATAC-Sequencing Method
Two commonly used methods for genome-wide detection of open chromatin are DNase-I hypersensitive sites sequencing and Assays for Transposase-Accessible Chromatin sequencing. DNase-seq and ATAC-seq are established on the use of cleavage enzymes (DNase-I and Tn5, respectively) that in open chromatin regions identify and cleave DNA. By distinguishing genomic intervals with many reads, sequencing, and the arrangement of reads from these fragments enables the identification of open chromatin. However, in an otherwise nucleosome-free area, the existence of transcription factors (TFs) linked to the DNA restricts the enzyme from cleavage. This gives small areas, known as footprints, where read scope suddenly decreases within heavy scope peak areas.
The Transposase-Accessible Chromatin assay accompanied by sequencing (ATAC-seq), a methodology for analyzing open chromatin regions, is especially interesting because, in small cell count samples, it is fast, simple, affordable, and portable. ATAC-seq features are usually built to distinguish open chromatin regions which, if these areas intertwine with protein-binding sites, can be employed to deduce TF binding occurrences. In relation, recent research has appeared that TF action can be informed by adjustments in chromatin availability. In particular, BagFoot coupled footprinting with differential availability to define, in the existence of a disturbance, TFs related with altered chromatin accessibility profiles. They concentrated primarily on hypersensitivity information from DNase I, but also analyzed a tiny proportion of datasets from ATAC-seq.
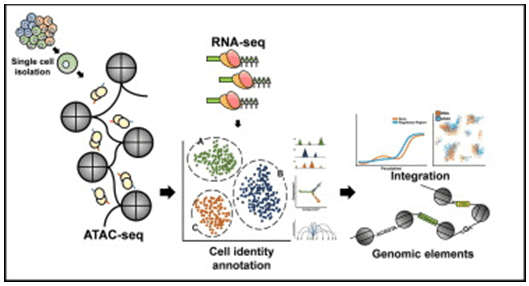
Figure 1. Single-cell ATAC sequencing analysis. (Baek, 2020)
Depending on either bulk ATACseq data or a combination of single-cell ATAC-seq (scATAC-seq) data as bulk data, a previously published model, HINT-ATAC, has been developed to evaluate TF binding at the cell population level. Deep learning methods have become a potent technique for analyzing TF binding structures in recent years, such as coevolutionary neural networks (CNNs). Techniques such as FactorNet and deep ATA leverage deep learning-based methods to define open chromatin areas and use bulk chromatin accessibility information to derive TF binding areas. All these techniques, however, make population-level TF binding assumptions and thus do not take heterogeneity into account within cellular communities.
Recent advancements in single-cell epigenomic sequencing allow a single-cell level of chromatin availability to be characterized. For instance, scATAC-seq has become feasible to test chromatin availability within single cells, allowing the recognition of cis- and trans-regulators and the test of how these regulators cooperate to affect cell fate in various cells. Employing only scATAC-seq info is difficult, as in all single-cell sequencing technologies, because they are limited and loud due not only to technical restrictions such as shallow sequencing but also to biological actualities like cellular uniformity.
References
- Fu L, Zhang L, Dollinger E, et al. Predicting transcription factor binding in single cells through deep learning. Science Advances. 2020 Dec 18;6(51).
- Baek S, Lee I. Single-cell ATAC sequencing analysis: From data preprocessing to hypothesis generation. Computational and structural biotechnology journal. 2020 Jan 1;18.
- Tripodi IJ, Allen MA, Dowell RD. Detecting differential transcription factor activity from ATAC-seq data. Molecules. 2018 May;23(5).