
 Sample Submission Guidelines
Sample Submission Guidelines
Introduction to Single Cell Genomics
James Eberwine et al. and Iscove and colleagues were the first to sequence an entire transcriptome at the level of a single cell, using linear amplification by in vitro transcription and exponential amplification by PCR, respectively, to expand the complementary DNAs (cDNAs) of a single cell. These methods were first employed on commercially available high-density DNA microarray chips, and then adjusted for single-cell RNA sequencing (scRNA-seq). In 2009, the first description of single-cell transcriptome analysis using a next-generation sequencing platform was published, and it described the characterization of cells at different stages of development. Since the publication of this paper, there has been a surge in interest in obtaining global high-resolution views of single-cell heterogeneity. Importantly, analyzing gene expression differences between individual cells has the potential to identify rare populations that would otherwise go undetected in a pooled cell analysis. The ability to identify and characterize outlier cells within a population, for example, could help us better understand drug resistance and relapse in cancer treatment. Researchers have recently been able to deconvolute highly diverse immune cell populations in healthy and diseased states thanks to significant advances in available experimental techniques and bioinformatics pipelines. scRNA-seq is also increasingly being used to determine cell lineage relationships in early development, myoblast differentiation, and lymphocyte fate determination.
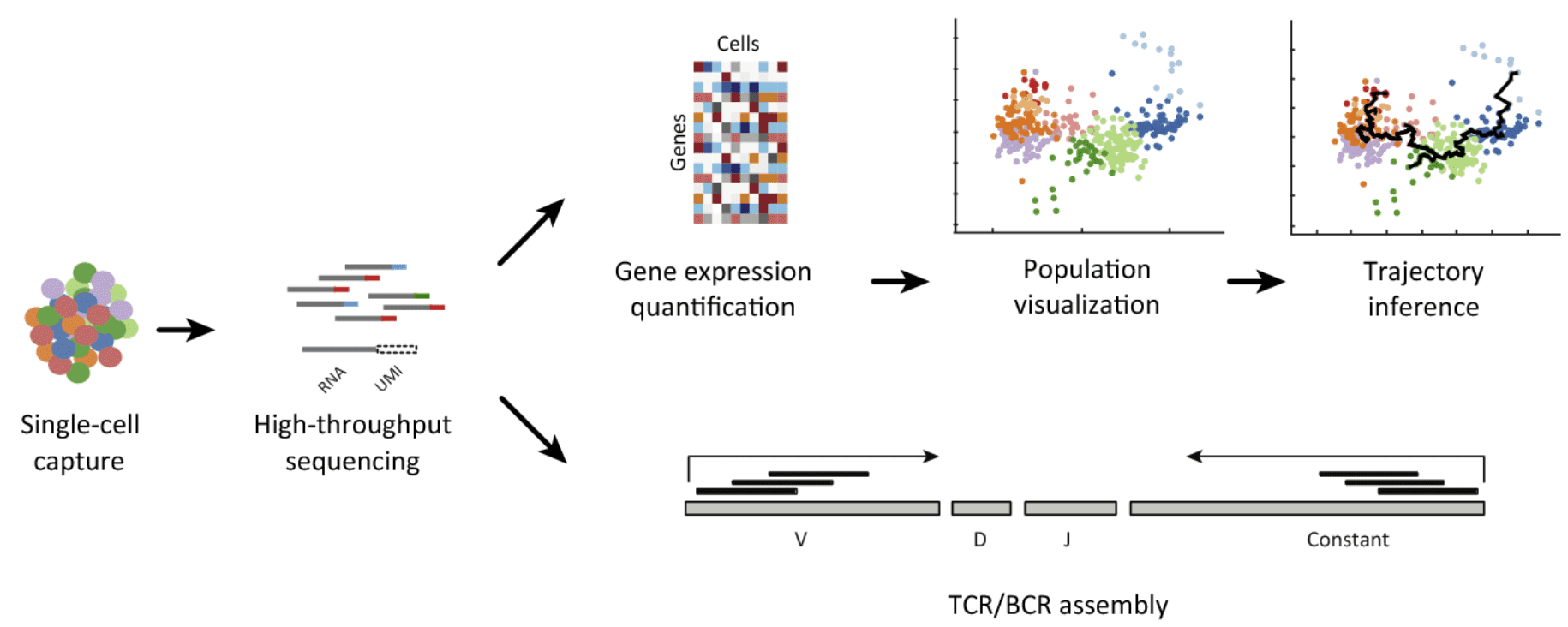
Bioinformatics In Single Cell Genomics
Because only a small number of cells are usually available for analysis in developmental biology, single-cell analysis becomes especially important, as minute spatial and temporal differences can lead to huge changes in cell behavior due to the inherent 4 differentiation process. The experimental phase of single-cell analysis is divided into three stages: (i) biotechnological and microfluidics approaches; (ii) imaging, sequencing, microarray, spectrometry, and other platforms for data acquisition; and (iii) data analysis.
Imaging: Since the invention of the microscope, much of the progress in cell biology has come from viewing proteins and cellular components. Despite issues with its reliability, antibody staining has been the standard method for observing proteins in fixed cells.
Data Analysis- Normalization: The need for normalization due to amplification biases presented by limited quantities of starting RNA/DNA material is one of the first issues in single-cell bioinformatics analysis. This problem should be solved by combining experimental and computational approaches.
Comparative Analysis: The genomic and/or transcriptomic information on dozens of individual cells is acquired in a single-cell sequencing project. The goal of comparative studies is to find structural variants or transcripts that are plentiful in different cells or cell populations.
Low Dimensional Analysis: The genomic and/or transcriptomic information on dozens of individual cells is acquired in a single-cell sequencing project. The goal of comparative studies is to find structural variants or transcripts that are plentiful in different cells or cell populations.
Applications of Single Cell Genomics
The technique of scRNA-seq is revolutionizing our basic understanding of biology, and it has opened up new research frontiers that go beyond descriptive studies of cell states. This technology could be used in a variety of exciting medical applications. Tumor heterogeneity is a common occurrence that can occur both within and between tumors, and we expect that scRNA-seq will be able to shed light on previously unknown tumor features that have been missed by bulk transcriptomic studies. In this way, scRNA-seq could aid in the development of cancer evolution models. By modeling transcriptional kinetics, this method could also be used to rebuild clonal and phylogenetic relationships between cells.
References:
- Yang J, Liao B, Zhang T, Xu Y. Bioinformatics analysis of single cell sequencing data and applications in precision medicine. Frontiers in genetics. 2020 Jan 23;10.
- Inda MC, Joshi S, Wang T, et al. The epichaperome is a mediator of toxic hippocampal stress and leads to protein connectivity-based dysfunction. Nature communications. 2020 Jan 16;11(1).
- Neu KE, Tang Q, Wilson PC, Khan AA. Single-cell genomics: approaches and utility in immunology. Trends in immunology. 2017 Feb 1;38(2).
- Eberwine J, Yeh H, Miyashiro K, et al. Analysis of gene expression in single live neurons. Proceedings of the National Academy of Sciences. 1992 Apr 1;89(7).