
 Sample Submission Guidelines
Sample Submission Guidelines
DNA methylation, histone variant, histone modifications, and nucleosome positioning are pathways for epigenetic regulation in eukaryotes. Bacteria, on the other hand, lack histones and nucleosomes; hence, they primarily use DNA methylation as their epigenetic gene regulation. There are three different types of DNA methylation in bacterial genomes: N4-methylcytosine (4mC), N6-methyladenine (6mA), and 5-methylcytosine (5mC). The methyltransferase (MTase) enzyme methylates DNA by transferring a methyl group from S- adenosyl-l- methionine (SAM) on target bases. Only a few sequence motifs in each bacterial genome are targeted by MTases. In Escherichia coli, 5ʹ-GATC-3ʹ is targeted by DNA adenine methylase (Dam) and 5ʹ-CCWGG-3ʹ by DNA- cytosine methyltransferase (Dcm). The large diversity of methylated motifs across the bacterial kingdom resulted from the great variation of the MTase specificity domain.
DNA methylation takes part in vital pathways in the biology of bacteria such as partitioning chromosomes to daughter cells, the timing of DNA replication, DNA repair, and timing of transposition and conjugal transfer of plasmids that are sensitive to the states of specific DNA region methylation. In bacteria, most epigenetic systems use DNA methylation to regulate a specific DNA-protein interaction. The system comprises a DNA methylase and a DNA binding protein(s) that bind to DNA sequences overlapping the methylation site, hence blocking methylation of that target site. In turn, methylation of the target site inhibits protein binding, producing two alternative methylation states of the target site – methylated and nonmethylated.
The detailed sequence targets and roles of most MTases remain unknown which can be attributed to the lack of high-throughput tools and methods for identifying 6mA and 4mC types. While development for methods such as detecting eukaryotic 5mC has relatively flourished, minor advances for methods facilitating the identification of DNA methylation forms in prokaryotic (bacterial) genomes have been made over the same period of time. Recently, new epigenomics sequencing technologies are beginning to address this problem, as shown in Table 1. Specifically, single-molecule, real-time, or SMRT sequencing has enabled all three major bacterial DNA methylation forms to be identified simultaneously for the first time but in different sensitivities depending on the form of methylation.
Table 1. DNA sequencing methods currently used for DNA methylation detection in prokaryotes (Beaulaurier et al., 2019).
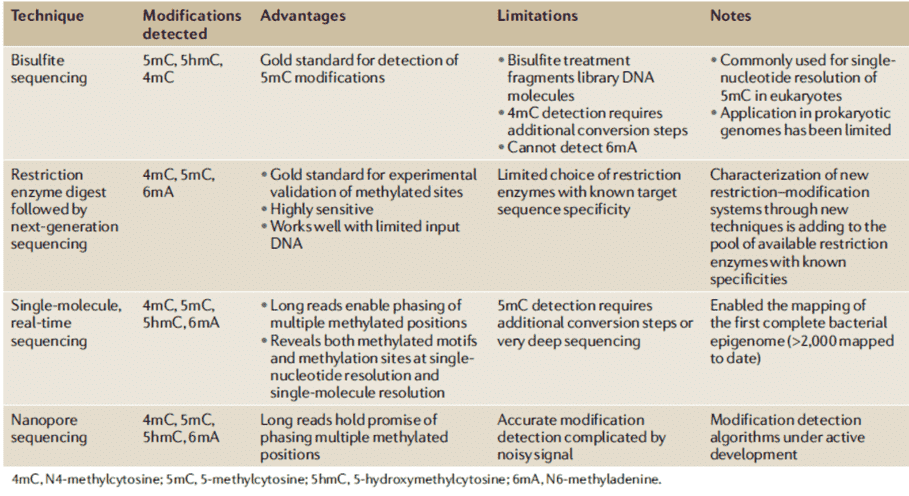
Comprehensive mapping, as detailed in Figure 1, of the bacterial methylome requires the inclusion of motifs and MTases. For all three of the methylation types, occurrences of DNA methylation in bacterial genomes are highly motif-driven. In identifying methylation motifs, it is important to note that most of the time, >95% of motif events are methylated if a motif is targeted by an MTase. For the identification of MTases producing motif methylation, tools for homology search and gene prediction, such as SEQWARE, facilitate the identification of genes having a high probability to encode RM system components such as subunits responsible for restriction, MTase activity, and specificity. RM systems often lead to low transformation efficiencies. As a solution, the effective design of appropriate shuttle vectors must include methylation patterns to provide optimum methylation or reduction of motif sites in the vector that are vulnerable to restriction by the host RM system.
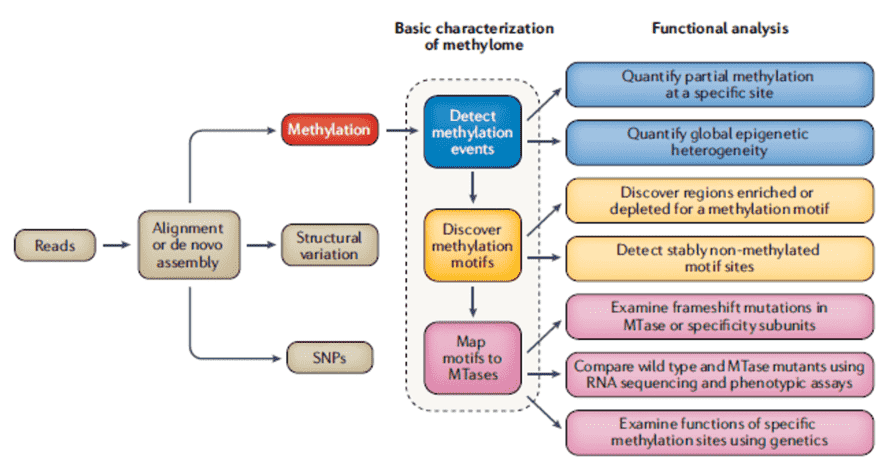
Figure 1. Comprehensive steps for methylome characterization in bacteria (Beaulaurier et al., 2019)
References
- Beaulaurier J, Schadt EE, Fang G. Deciphering bacterial epigenomes using modern sequencing technologies. Nature Reviews Genetics. 2019, 20(3).
- Casadesús J, Low D. Epigenetic gene regulation in the bacterial world. Microbiology and molecular biology reviews. 2006, 70(3).