
 Sample Submission Guidelines
Sample Submission Guidelines
Variant Calling: From Sequence Data to Reliable Mutation Detection
Variant calling is a pivotal process in genomic research, enabling the identification of genetic differences between an individual's genome and a reference genome. This article provides a comprehensive overview of the best practices for variant calling in clinical sequencing, covering data preprocessing, alignment, variant detection algorithms, filtering, and validation. It also discusses the challenges and future directions in this rapidly evolving field, emphasizing the importance of rigorous methods and benchmarking to ensure reliable mutation detection.
What is Variant Calling
Identifying genomic variations represents a critical component in genomics research, aimed at detecting genomic differences between individual samples and reference sequences through analysis of sequencing information. The spectrum of these variations encompasses several categories: single nucleotide polymorphisms (SNPs), insertion and deletion events (indels), larger structural rearrangements (SVs), and additional variant classifications that potentially impact biological pathways or contribute to pathogenesis.
The process of identifying these genetic differences holds substantial importance across genomic studies and serves a fundamental function in numerous scientific and clinical domains. Within medical practice, the detection of genomic variations facilitates the identification of pathogenic mutations, thereby establishing crucial foundations for disease screening programs, precision medicine approaches, and genetic counseling services.
Services you may interested in
Data Preprocessing and Read Alignment of Variant Calling
Quality control optimization of raw sequencing data
With the gradual maturity of second-generation sequencing technology and the continuous expansion of its application scope, it is particularly important to clarify the standards of data quality, reliability, repeatability and biological relevance. Data quality plays a vital role in various downstream analyses such as sequence assembly, SNP identification and gene expression studies. Therefore, it is crucial to perform quality control on the raw sequencing data before data analysis. FastQC is a widely used quality assessment tool that can quickly generate a quality report for sequencing data. It evaluates data quality from multiple dimensions, such as base quality distribution, GC content distribution, sequence repetition rate, etc.
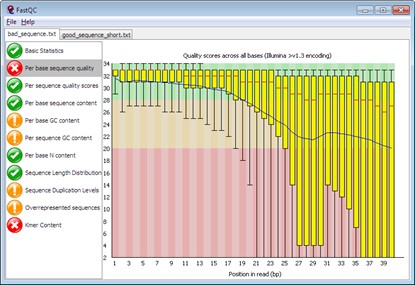 Figure 1. FastQC report.(From FastQC website)
Figure 1. FastQC report.(From FastQC website)
After completing the quality assessment, the data needs to be filtered. Trimmomatic (Bolger et al. 2014) and BBDuk are commonly used filtering tools. Trimmomatic can perform various processing on sequencing data, including removing adapter sequences, trimming low-quality bases, etc. BBDuk also has powerful filtering functions, which can efficiently identify and remove adapter sequences. When identifying adapter sequences, BBDuk will compare the sequencing data with known adapter sequences and accurately identify the adapters by setting appropriate matching thresholds. For the trimming of low-quality bases, BBDuk can dynamically adjust according to the base quality value.
Core technologies and tool selection for sequence alignment
Sequence alignment is a key step in variant detection, which locates variant sites by matching short sequencing reads to the reference genome. Common alignment tools such as BWA (based on seed extension, suitable for long reads and complex genomes) and Bowtie2 (based on BWT transformation, suitable for short reads and fast alignment) use different algorithms to meet different needs, while STAR is specifically designed for RNA-seq data and can effectively handle splicing alignment.
Local alignment and global alignment are two different alignment strategies, applicable to different scenarios. Local alignment only considers the similar parts of the sequence, and does not require the entire sequence to match completely. It is often used to find conserved regions in the sequence or detect mutations. Global alignment requires the entire sequence to be aligned, which is suitable for comparing two similar sequences. For example, when detecting SNPs, local alignment can more accurately find the mutation site; and when comparing homologous genes of two species, global alignment is more appropriate.
Variant Calling Algorithms and Tools
Analysis of traditional probability model method
One of the commonly used tools for variant detection is the HaplotypeCaller module in the GATK software. This module estimates the gene combinations of different haplotypes and calculates the probabilities of each combination. Based on these probabilities, the reverse reasoning method is used to determine the genotype of each sample. The HaplotypeCaller module is not only suitable for population variant detection, but also can infer individual mutation information and genotype distribution based on population information.
Bayesian statistical models are widely used in GATK HaplotypeCaller. The model takes into account multiple factors, such as sequencing error rate, base quality value, etc., to calculate the probability that each site is a variant site.In this way, the authenticity of the variant can be judged more accurately and false positive results can be reduced.
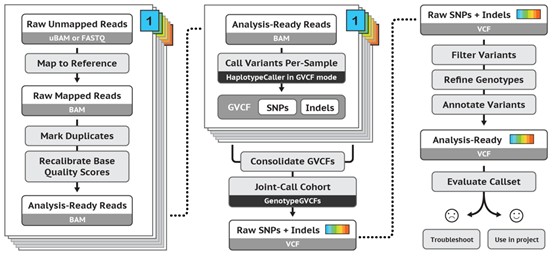 Figure 2. GATK work pipeline.(From GATK website)
Figure 2. GATK work pipeline.(From GATK website)
The basic workflow of SNP/indel detection using the GATK-HaplotypeCaller module consists of four main steps:
- 1)Identifying active areas
- 2)Determination of haplotypes by reassembly of active regions
- 3) Determine the likelihood value of the haplotype for each read
- 4) Determine genotype
Slide along the reference genome in a certain window, calculate the activity score of each position in the genome by statistically comparing mismatches, indels and softclips, and use the smoothing algorithm for processing, which is equivalent to measuring the entropy value of the region. When the entropy value reaches a certain set threshold, the region is determined to be an active region for subsequent assembly.
For each active region, the previous read alignment results are ignored and the reads in the region are reused to build a De Bruijn-like graph to assemble active regions and identify possible haplotypes in the data. Then, the Smith-Waterman algorithm is used to realign each haplotype with the reference haplotype to identify potential variant sites.
For each active region, the program uses the PairHMM algorithm to align each read with each haplotype, generating a matrix of haplotype likelihood values. These likelihood values are then marginalized to obtain the allele likelihood for each potential variant site for a given read.
The likelihood values of candidate haplotypes obtained in the previous PairHMM step are converted into the likelihood values of the genotypes at each site using the Bayesian algorithm .
In addition to GATK HaplotypeCaller, there are also tools such as Samtools and VarScan. Samtools is a powerful tool that can perform a variety of operations on sequencing data, including sorting, indexing, and variant detection. Its advantages are fast speed and ability to handle large-scale sequencing data; its disadvantage is that its accuracy is relatively low when dealing with complex variants. VarScan focuses on variant detection in tumor samples, and it can detect low-frequency somatic variants. However, VarScan may miss some rare variants when detecting them.
The detection revolution driven by deep learning
Deep learning has sparked a revolution in the field of variant detection, and DeepVariant is a typical example. Its core is the convolutional neural network (CNN) architecture, which can automatically learn characteristic patterns in sequencing data. CNN consists of multiple convolutional layers, pooling layers, and fully connected layers. The convolutional layer slides the convolution kernel over the input data to extract local features; the pooling layer reduces the dimension of the features to reduce the amount of calculation; the fully connected layer integrates the extracted features and outputs the final prediction results.
DeepVariant's error correction mechanism is a highlight. During the sequencing process, certain errors will occur due to various factors. DeepVariant can effectively identify and correct these errors by learning the characteristic differences between normal sequencing data and erroneous data through CNN. For example, it can identify base errors caused by sequencer errors and improve the accuracy of variant detection.
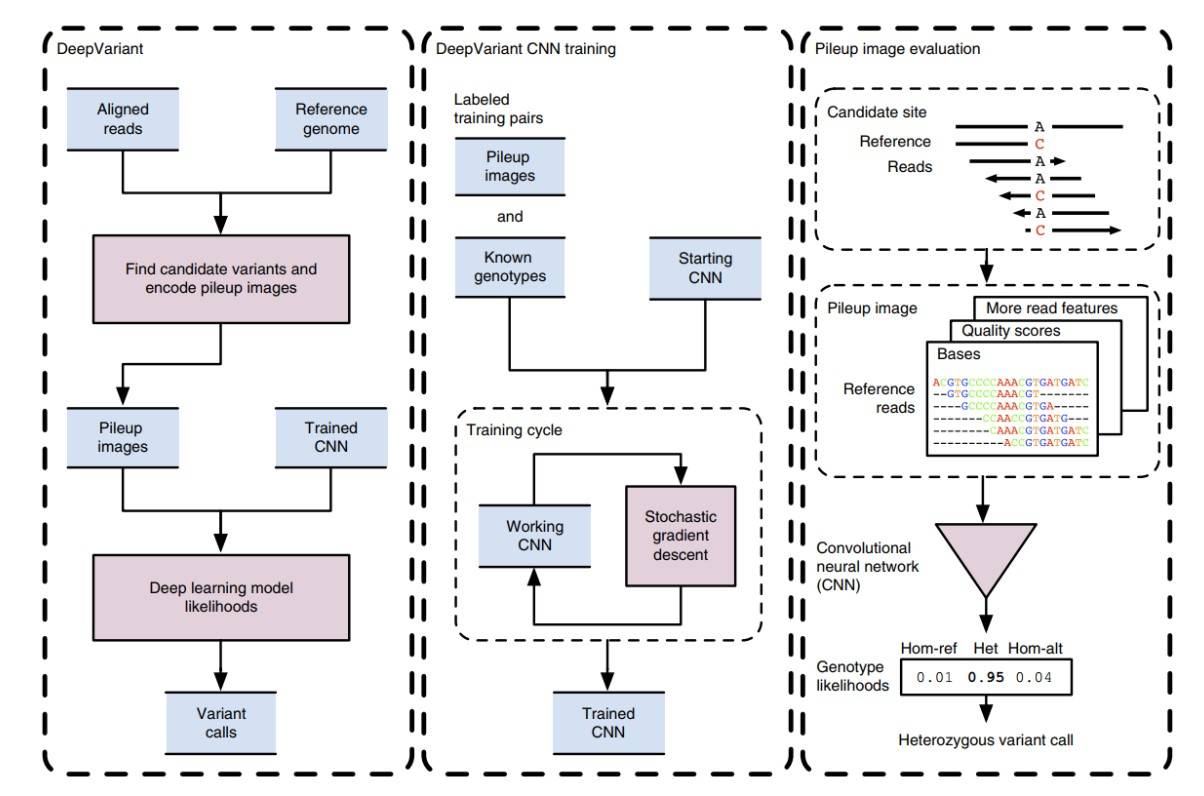 Figure 3. DeepVariant workflow overview.(Poplin, R. et al. 2018)
Figure 3. DeepVariant workflow overview.(Poplin, R. et al. 2018)
Filtering and Annotation ofVariant Calling
In variant detection, it is crucial to evaluate and screen the quality of variant results, which can effectively remove false positive variants and improve the reliability of test results. DP, QUAL, FS, etc. are commonly used quality assessment indicators.
DP (Depth) refers to sequencing depth, that is, the number of times a certain site is covered by sequencing. Generally speaking, the higher the sequencing depth, the higher the accuracy of variant detection. Usually, the DP threshold is set to 10-20, which means that a certain site is covered by sequencing at least 10-20 times before it is considered a reliable variant site. QUAL (Quality) is the quality score of the variant site, which comprehensively considers factors such as sequencing quality and alignment quality. The higher the QUAL value, the higher the credibility of the variant site. The common QUAL threshold can be set to 30. FS (Fisher Strand Bias) is used to detect whether there is a deviation in the distribution of variant sites on the positive and negative chains. If the FS value is too high, it may mean that the variant is caused by sequencing errors or alignment errors. The FS threshold is generally set to 20.
The multi-dimensional filtering strategy combines multiple quality assessment indicators for comprehensive screening. For example, considering DP, QUAL, and FS at the same time, a variant site is retained only when its DP is greater than 10, QUAL is greater than 30, and FS is less than 20. This strategy can more effectively remove false positive variants.
In tumor samples, VAF (Variant Allele Frequency) is an important parameter. Due to the heterogeneity of tumor cells, low-frequency somatic mutations may exist in tumor samples. The setting of the VAF threshold needs to be adjusted according to the specific situation. Generally speaking, for high-purity tumor samples, the VAF threshold can be set to 5% - 10%; for low-purity tumor samples, the VAF threshold may need to be reduced to 1% - 5%.
Challenges in Accurate Mutation Calling
Traditional sequencing technology is susceptible to sequencing errors and background noise when detecting low-abundance mutations, resulting in an increase in false positive and false negative results. For example, factors such as base errors and PCR amplification bias during sequencing can affect the accurate detection of low-abundance mutations. In addition, contamination during sample processing may also introduce false mutation signals, further reducing the accuracy of detection.
In precision detection, the analysis of complex genomic regions is a very challenging task, among which tandem repeats and GC preference are the main interfering factors. Tandem repeats refer to regions in the genome where short DNA sequences are repeated multiple times. The sequences in these regions have high similarity, which can easily lead to sequencing errors and alignment difficulties. GC preference refers to the high GC content in certain regions of the genome. Due to the strong hydrogen bonds between GC bases, amplification bias and signal attenuation are prone to occur during the sequencing process, affecting the accuracy of sequencing.
References:
- Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics(Oxford, England), 30(15), 2114–2120. https://doi.org/10.1093/bioinformatics/btu170
- Koboldt D. C. (2020). Best practices for variant calling in clinical sequencing. Genome medicine, 12(1), 91. https://doi.org/10.1186/s13073-020-00791-w


