
 Sample Submission Guidelines
Sample Submission Guidelines
Advances in Transcriptome Analysis Methods and Technologies
Transcriptomics is a subject that studies gene expression. By analyzing the transcriptome, people can understand which genes are activated, when and how their expression levels change. In this article, we first summarize and review common types of transcriptome technologies. Then we compare and analyze the technical advantages and main challenges of various research strategies to assist current experimental design and research analysis. Finally, we emphasize the importance of transcriptomics in multi-omics integrated analysis and look forward to its development prospects.
INTRODUCTION
Transcriptome, broadly speaking, refers to the sum of all RNAs that can be transcribed in a cell or a group of cells under the same environment (or physiological conditions), including messenger RNA (mRNA), ribosomal RNA (rRNA), transfer RNA (tRNA) and non-coding RNA[1]; in a narrow definition, it refers to all messenger RNA (mRNA) that can be transcribed by cells. Therefore, transcriptomics can be simply defined as follows: It is the study of all RNA molecules in cells. Transcriptomics, also known as expression profiling, studies the expression levels of RNA in a given cell population. Transcriptome analysis has a wide range of applications, including medicine, agriculture, environment and microorganisms[2-5]. Here we will start with transcriptome sequencing technologies and transcriptome analysis methods to explain the functional discovery of transcriptomics in the genome, reveal the molecular composition of cells and tissues, and understand the role of developmental and disease regulatory pathways.
TRANSCRIPTOME SEQUENCING TECHNOLOGIES
RNA-Seq is a recently developed technology that uses next-generation sequencing technologies for transcriptome analysis. It can comprehensively and quickly obtain the sequence information and expression information of almost all transcripts of a specific cell or tissue in a certain state, including protein-coding mRNA and various non-coding RNAs, the expression abundance of different transcripts produced by alternative splicing of genes, etc[6,7]. While analyzing the structure and expression level of transcripts, unknown transcripts and rare transcripts can also be discovered, thereby accurately analyzing important issues in life sciences such as gene expression differences, gene structural variations, and screening of molecular markers[8].
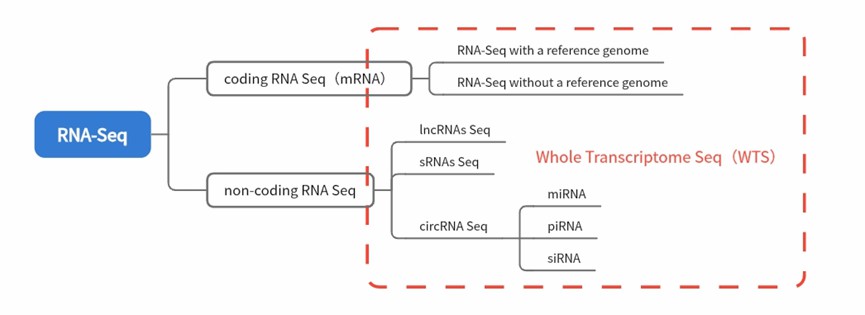 Fig.1 Classification of RNA-Seq technologies
Fig.1 Classification of RNA-Seq technologies
Service you may intersted in
Resource
The workflow for RNA sequencing is exactly similar (Fig. 2). It starts with sample quality control (Sample QC) to ensure that your samples meet the criteria of the RNA-Seq technique. Then, the appropriate library is prepared according to your target organism and application, and subsequently tested for its quality (Library QC). Next, a sequencing strategy is used to sequence the samples and the resulting data is also checked for its quality (Data QC). Finally, bioinformatic analyses are performed and results are provided. You can analyze the sequencing results according to your own experimental purposes.
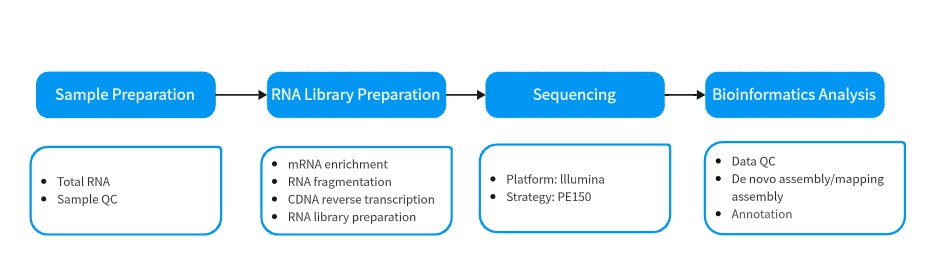 Fig.2 A typical RNA-seq experiment workflow
Fig.2 A typical RNA-seq experiment workflow
TRANSCRIPTOME ANALYSIS METHODS
Bulk Transcriptome Analysis
As mentioned above, the object of transcriptomic research is RNA under different or the same physiological/environmental conditions. They often have different functions. Transcriptomic analysis methods for RNA with different functions will be discussed below.
mRNAs are templates that guide the synthesis of proteins and are messengers that transmit genetic information from DNA to proteins. mRNA-seq is a powerful tool to analyze the cell transcriptome profile. Currently, there are many studies related to the mRNA transcriptome. The main research purposes/directions including:1.Quantitative profiling of transcripts in different tissues or samples, under various conditions and treatments.2.Discovery of novel transcripts, alternative splicing (AS), and transcript variations(Fig.3)[9].3.Research of developmental mechanisms[10] and drug resistance[11] through tissue-specific transcripts ortime-course gene expression(Fig.4).4.Biomarker discovery[12] based on novel transcripts/isoforms, SNP/InDel identification, and fusion geneanalysis.
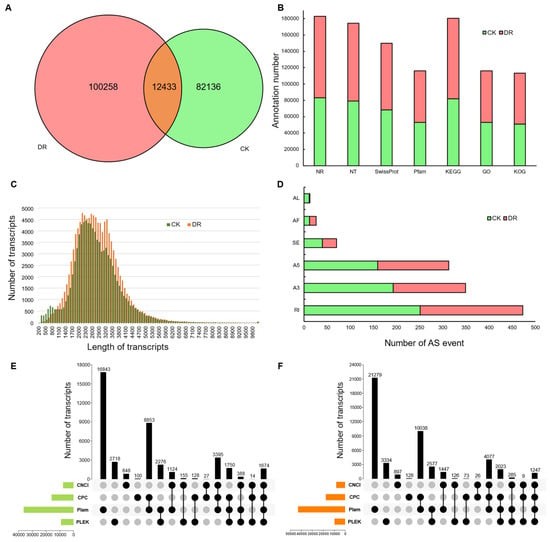 Fig.3 Gene function annotation and gene structure analysis of Lolium multiflorum
Fig.3 Gene function annotation and gene structure analysis of Lolium multiflorum
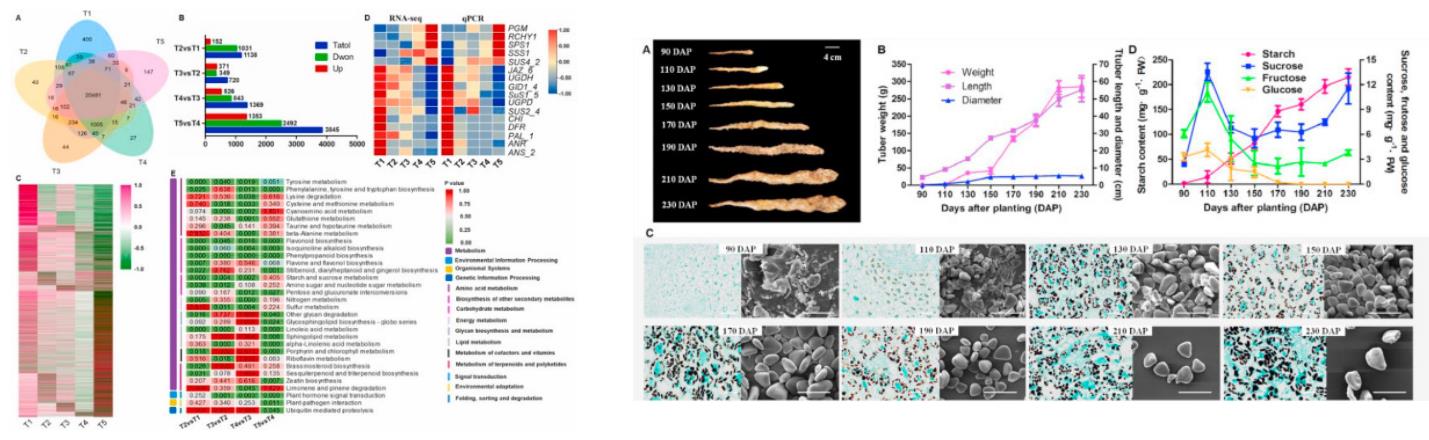 Fig.4 Transcriptome analysis of yam tubers at different developmental stages
Fig.4 Transcriptome analysis of yam tubers at different developmental stages
Long non-coding RNAs (lncRNAs) are a moderately abundant fraction of the eukaryotic transcriptome, which are comprised of longer than 200nt non-coding RNAs (ncRNAs) , which affect multiple cellular functions through their regulation of gene transcription, post-transcriptional modifications, and epigenetics. lncRNA sequencing (lncRNA-seq) is a powerful NGS tool to study functional roles in diverse biological processes and human diseases, such as cancer and neurological disorders[13,14].
Small RNAs (sRNAs) are short RNA molecules, usually non-coding, involved with gene silencing and the post-transcriptional regulation of gene expression. sRNA Sequencing (sRNA-seq) is a method that enables the in-depth investigation of these RNAs, in special microRNAs (miRNAs, 18-40nt in length). sRNA-seq is an effective approach to selectively target any species of sRNAs with unprecedented sensitivity and high resolution, all in a single analysis. Coupled with a robust in-house bioinformatics pipeline, sRNA-seq service has been assisting researchers to describe the differential expression of miRNAs, structural alterations, and to discover novel sRNAs[14,15].
Circular RNA (circRNA) is a highly stable molecule of ncRNA, in form of a covalently closed loop that lacks the 5'end caps and the 3' poly(A) tails. The circular structure grants circRNAs resistance against exonuclease digestion, a characteristic that can be exploited in library construction. CD Genomics's circRNA sequencing service (circRNA-seq) uses next-generation sequencing (NGS) technology to support a wide range of investigations focused on circRNA. The regulatory function of circRNAs may be involved in many biological processes[14], such as regulation of gene expression by acting as miRNA "sponges", transport of miRNAs, and overall regulation of protein synthesis.
Service you may intersted in
Resource
Transcriptome analysis based on Single-Cell Sequencing
There may be many cells with different phenotypes and genotypes in the same type of cells. single-cell RNA-seq technology provides scientists with the opportunity to study and analyze the behavior, mechanism, and relationship of individual cells by sequencing the transcriptome at the single-cell level[16]. It is widely used in hot research fields such as germ cells, embryonic stem cells, nerve cells, tumor cells, and immune cells. The current single-cell transcriptome sequencing platform 10x Genomics can capture nearly 100,000 cells using technologies such as microfluidics and barcode labeling to obtain a large amount of transcriptome information[17]. The core of single-cell transcriptome analysis is the classification of cells and the identification of differential genes in subpopulations. Grouping and differential analysis of cells are performed, and functional enrichment of differential genes is performed to identify the functional characteristics of subpopulations(Fig.5)[18]. It has unique advantages in cell typing and identification of marker factors, bringing new methods and convenient means for cell type identification, differential response of signaling pathways, molecular regulatory networks, and cell heterogeneity research[19,20].
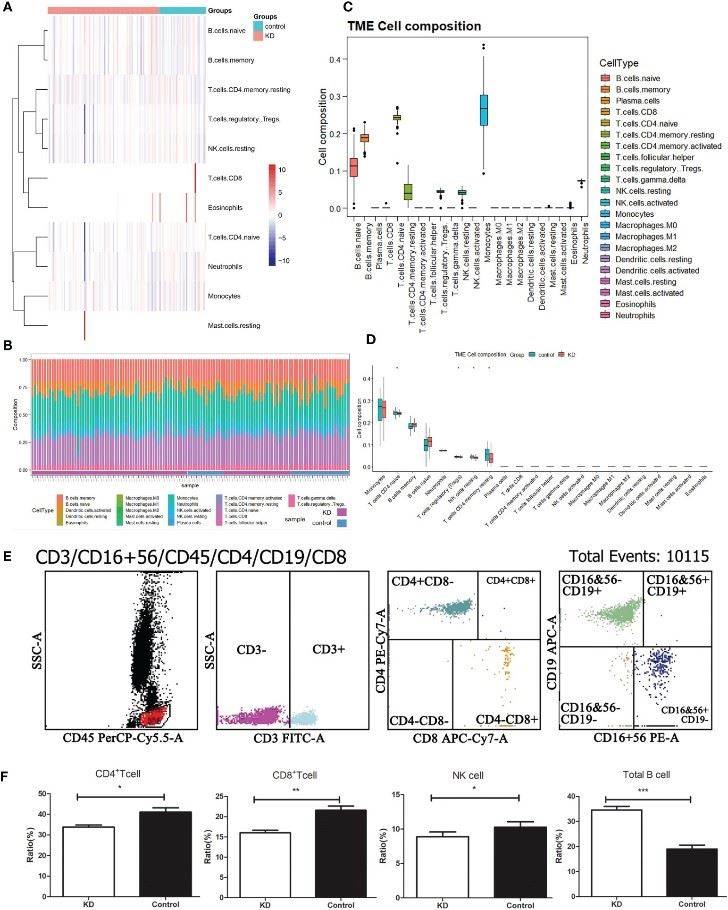 Fig.5 The landscape analysis of 22 types of immune cells from normal and Kawasaki disease patients.
Fig.5 The landscape analysis of 22 types of immune cells from normal and Kawasaki disease patients.
Transcriptome analysis based on Spatial Transcriptome Sequencing
Spatial transcriptome sequencing can provide data information such as the transcriptome of the research object, and can also locate its spatial position in the tissue. Combining the transcriptome information with the spatial position information provides a basis for detecting the spatial composition and gene expression of cells in the tissue[21,22]. Visium spatial transcriptome is a technology that detects the gene expression of the whole transcriptome in situ in tissues, which allows us to detect the gene expression levels and at the same time obtain the location information of the spatial expression of genes within the tissue. Compared with spatial transcriptome, traditional whole-gene transcriptome or single-cell transcriptome sequencing loses the spatial distribution information of genes or cells within tissues, while the widely used RNA probe hybridization can only detect a limited number of samples at the same time. Spatial transcriptome can superimpose gene expression and HE staining results without digesting the tissue(Fig. 6), thereby retaining the spatial structure within the tissue, allowing us to study the cellular heterogeneity in different regions within the tissue[23-25].
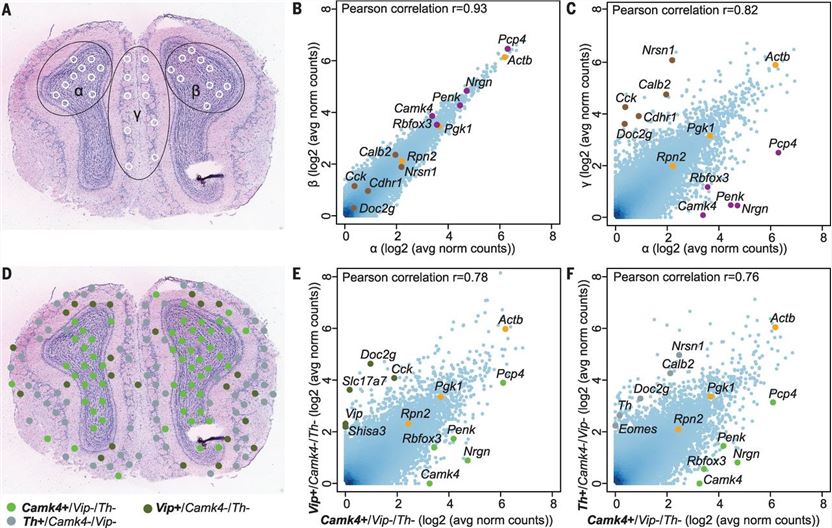 Fig.6 Visualization and bioinformatics analyses of tissue domains defined by morphology or gene expression profile.
Fig.6 Visualization and bioinformatics analyses of tissue domains defined by morphology or gene expression profile.
CONCLUSION AND FUTUREDIRECTIONS
In this article, we discussed several common strategies and sequencing technologies for transcriptome analysis. Bulk transcriptome sequencing can only detect the average expression level of genes in mixed cells and does not include specific cell and spatial location information[26]; single-cell transcriptome sequencing can obtain individual gene expression profiles of cells and explore cell heterogeneity, but the source of spatial location information cannot be obtained[16]; the spatial transcriptome provides spatial distribution information of gene expression profiles, thereby simultaneously obtaining cell gene expression data and spatial location information, and exploring spatial heterogeneity. It further promotes the study of real gene expression of tissue in situ cells and the morphological display of intercellular communication networks[22].
In the process of biomolecule analysis, it is an inevitable trend to go from the whole to the details, and then from the details back to the whole. When people understand the biological structure from the molecular and cellular level, they will naturally want to study cell groups and tissues at a more macro scale , and even the physiological processes and pathological connections of organs. Multi-omics research methods are not only widely used in the field of disease, but can also provide a comprehensive and systematic understanding of the interrelationships and regulatory networks of various biomolecules in the fields of basic research, molecular breeding, clinical diagnosis, and drug development, providing a basis for network biology and systems[27-29]. Therefore, the strategy of combining transcriptome with genome, proteome, metabolome and other multi-omics joint analysis is being adopted by more and more scientists. It is believed that multi-omics research strategies can open up more research fields in the future.
References:
- Hrdlickova R, Toloue M, Tian B. RNA-Seq methods for transcriptome analysis. WIREs RNA, 2017, 8(1): e1364.
- Hong M, Tao S, Zhang L, et al. RNA sequencing: new technologies and applications in cancer research. Journal of Hematology & Oncology, 2020, 13(1): 166.
- Li K, Chen X, Zhang J, et al. Transcriptome Analysis of Stephania tetrandra and Characterization of Norcoclaurine-6-O-Methyltransferase Involved in Benzylisoquinoline Alkaloid Biosynthesis. Frontiers in Plant Science, 2022, 13.
- Liu G, Bi X, Tao C, et al. Comparative transcriptomics analysis of Zygosaccharomyces mellis under high-glucose stress. Food Science and Human Wellness, 2020.
- Pan Y, Wang Y, Hao W, et al. Metatranscriptomics Unravel Composition, Drivers, and Functions of the Active Microorganisms in Light-Flavor Liquor Fermentation. Microbiol Spectr, 2022, 10(3): e0215121.
- Soneson C, Delorenzi M. A comparison of methods for differential expression analysis of RNA-seq data. BMC Bioinformatics, 2013, 14(1): 91.
- Marguerat S, Bähler J. RNA-seq: from technology to biology. Cell Mol Life Sci, 2010, 67(4): 569-79.
- Conesa A, Madrigal P, Tarazona S, et al. A survey of best practices for RNA-seq data analysis. Genome Biology, 2016, 17(1): 13.
- Liu Q, Wang F, Shuai Y, et al. Integrated Analysis of Single-Molecule Real-Time Sequencing and Next-Generation Sequencing Eveals Insights into Drought Tolerance Mechanism of Lolium multiflorum. International Journal of Molecular Sciences, 2022, 23(14): 7921.
- Cao T, Wang S, Ali A, et al. Transcriptome and metabolome analysis reveals the potential mechanism of tuber dynamic development in yam (Dioscorea polystachya Turcz.). LWT, 2023, 181: 114764.
- Ruiz De Garibay G, Roch B, Garrido López P, et al. 1010P Assessment of early resistance mechanisms to first-line osimertinib in EGFR-mutant NSCLC using spatial transcriptomics. Annals of Oncology, 2022.
- Liu W, Qiu W, Huang Z, et al. Identification of nine signature proteins involved in periodontitis by integrated analysis of TMT proteomics and transcriptomics. Frontiers in Immunology, 2022, 13.
- Goede O M D, Nachun D C, Ferraro N M, et al. Population-scale tissue transcriptomics maps long non-coding RNAs to complex disease. Cell, 2021, 184(D1).
- Li J W, Ping. Transcriptomic analysis of high-throughput sequencing about circRNA, lncRNA and mRNA in bladder cancer. Gene: An International Journal Focusing on Gene Cloning and Gene Structure and Function, 2018, 677.
- Koeppen K, Hampton T H, Jarek M, et al. A Novel Mechanism of Host-Pathogen Interaction through sRNA in Bacterial Outer Membrane Vesicles. PLoS Pathogens, 2016, 12(6): e1005672.
- Chen G, Ning B, Shi T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front Genet, 2019, 10: 317.
- Gao C, Zhang M, Chen L. The Comparison of Two Single-cell Sequencing Platforms: BD Rhapsody and 10x Genomics Chromium. Curr Genomics, 2020, 21(8): 602-609.
- Shaw R, Tian X, Xu J. Single-Cell Transcriptome Analysis in Plants: Advances and Challenges. Mol Plant, 2021, 14(1): 115-126.
- Cao N, Ouyang H, Zhang X, et al. Integration of scRNA-Seq and bulk RNA-Seq uncover perturbed immune cell types and pathways of Kawasaki disease. Frontiers in Immunology, 2023, 14.
- Nahrendorf M, Swirski F K. Abandoning M1/M2 for a Network Model of Macrophage Function. Circulation Research, 2016, 119(3): 414-417.
- Ståhl P L, Salmén F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science, 2016, 353(6294): 78-82.
- Du J, An Z J, Huang Z F, et al. Novel insights from spatial transcriptome analysis in solid tumors. Int J Biol Sci, 2023, 19(15): 4778-4792.
- Jia E, Sheng Y, Shi H, et al. Spatial Transcriptome Profiling of Mouse Hippocampal Single Cell Microzone in Parkinson's Disease. Int J Mol Sci, 2023, 24(3).
- Rao A, Barkley D, França G S, et al. Exploring tissue architecture using spatial transcriptomics. Nature, 2021, 596(7871): 211-220.
- Wang Y, Liu B, Zhao G, et al. Spatial transcriptomics: Technologies, applications and experimental considerations. Genomics, 2023, 115(5): 110671.
- Bivens N J, Zhou M. RNA-Seq Library Construction Methods for Transcriptome Analysis. Current Protocols in Plant Biology, 2016, 1(1): 197-215.
- Li D, Lu X, Zhu Y, et al. The multi-omics basis of potato heterosis. J Integr Plant Biol, 2022, 64(3): 671-687.
- Wang J M, Hong R, Demicco E G, et al. Deep learning integrates histopathology and proteogenomics at a pan-cancer level. Cell Reports Medicine, 2023, 4(9).
- Sveinbjornsson G, Ulfarsson M O, Thorolfsdottir R B, et al. Multiomics study of nonalcoholic fatty liver disease. Nature Genetics, 2022, 54(11): 1652-1663.


