
 Sample Submission Guidelines
Sample Submission Guidelines
Workflow of LncRNA Sequencing and Its Data Analysis
Introduction to LncRNA sequencing
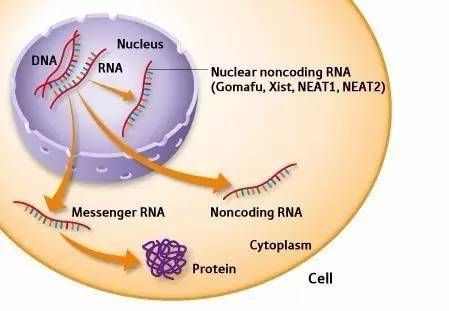
LncRNA is non-coding RNA with a length of more than 200 nucleotides. Compared with mRNA, lncRNA is generally low in expression and has stronger tissue specificity. As a hot spot of RNA research, lncRNA regulates the expression of coding genes at various levels, including epigenetic inheritance, transcription and post-transcription. Different from traditional chip inspection, lncRNA analysis combined with high-throughput sequencing technology and bioinformatics analysis can comprehensively excavate the information of lncRNA in samples. LncRNA sequencing technology has been widely used in genetic improvement of species, and disease studies on occurrence, development, diagnosis, and treatment.
Long-noncoding RNA (lncRNA) sequencing involves high-throughput sequencing of a class of non-coding RNAs produced within cells with a length exceeding 200 nucleotides. Library construction is accomplished using a method that eliminates ribosomal RNA (rRNA), followed by deploying high-throughput sequencing technologies, backed by a robust bioinformatics analysis platform. This approach allows for comprehensive and in-depth studies of all known or novel lncRNAs present within a given sample.
Utilizing strand-specific library construction for lncRNAs imbues the approach with certain advantages relative to conventional transcriptome library construction techniques. Primary among these is that strand-specific sequencing can establish the transcriptional directionality of the two RNA strands, thereby reducing errors during the alignment process. Furthermore, lncRNA library sequencing yields richer information. Constructing an lncRNA library is carried out in the purview of mRNA + lncRNA, such that a single round of library construction simultaneously results in data pertaining to both mRNA and lncRNAs.
Services you may interested in
The Workflow of LncRNA Sequencing
A typical lncRNA sequencing includes the quality assessment of total RNA, library preparation and sequencing. From the RNA sample to the final data, every step has an impact on the data quality and quantity. Therefore, obtaining high-quality data is the premise of ensuring comprehensive and credible biological information analysis.
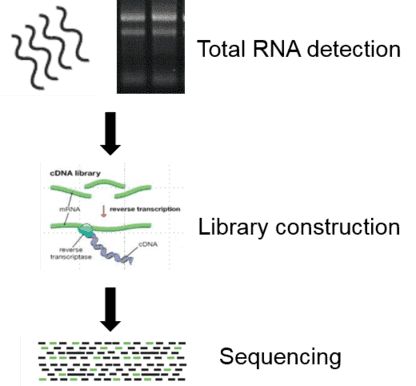 Figure 1. Overview of lncRNA sequencing.
Figure 1. Overview of lncRNA sequencing.
Total RNA detection: It mainly includes analysis of RNA degradation degree and contamination, detection of RNA purity (OD260/280 ratio), accurate quantification of RNA concentration (Qubit) and RNA integrity detection.
Library construction: After the total RNA is qualified for library preparation, the poly-A based mRNA enrichment followed by mRNA fragmentation is performed to reduce rRNA reads. Then the cDNA is synthesized from enriched and fragmented RNA using reverse transcriptase (Super-Script II) and random primers. The cDNA was further converted into double-stranded DNA using the buffer solution, dNTPs (dUTP, dATP, dGTP and dCTP) and polymerase. The cDNA fragments are repaired at the end and ligated to platform-specific adapters.
The key difference between lncRNA-seq and conventional transcriptome sequencing library construction lies in the method of target RNA enrichment. The former employs a negative selection process to remove rRNA, while the latter uses positive selection to enrich mainly mRNA with subsequent steps being identical. Upon sample qualification, library construction is initiated. Total RNA (>200) is extracted from tissue samples and rRNA is removed from the sample using a reagent kit. Following fragmentation, random primers and reverse transcriptase are used to synthesize the first cDNA strand, followed by second-strand cDNA synthesis. Purified double-stranded cDNA undergoes end repair, adenylate addition, and sequencing adaptor ligation. Finally, cDNA libraries are obtained through PCR amplification.
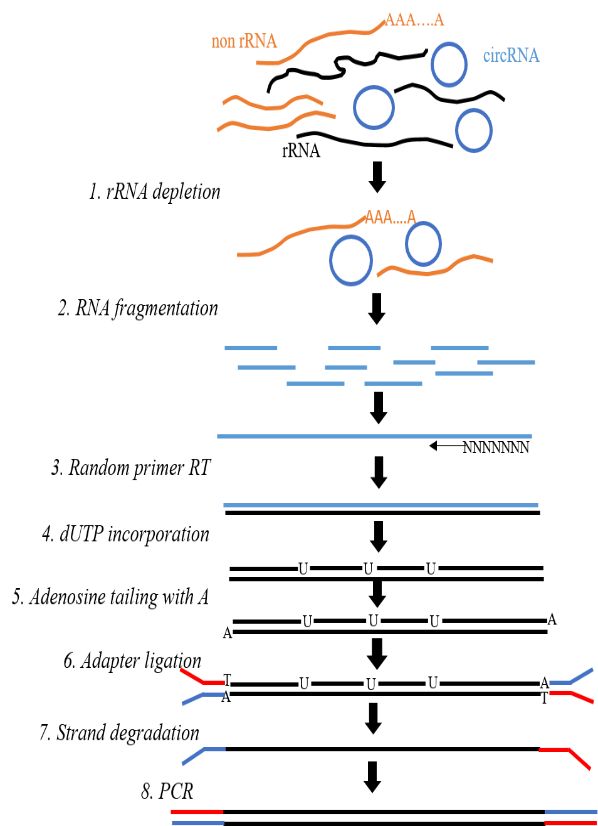 Figure 2. Illustration of lncRNA-seq library construction.
Figure 2. Illustration of lncRNA-seq library construction.
Library detection and sequencing: After the library construction, initial quantification and dilution are carried out, and then the fragment size of library is tested. To determine the quality of the library, we can look primarily at the degradation status of the mRNA and the size of the inserted fragments. If mRNA degradation occurs, sequences that have degraded will show few, or even no read alignments. The higher the randomness of the mRNA fragmentation, the more uniform the coverage of reads within each area. Hiseq/Miseq sequencing is performed following the library inspection.
LncRNA Sequencing Data Analysis
The analysis process for lncRNA-seq, as compared to transcriptome sequencing, embodies a significant distinction — the need to discriminate between mRNA and lncRNA. The fundamental difference between mRNA and lncRNA is the non-coding nature of lncRNA. Based on their coding potential, a differentiation can be made between mRNA and lncRNA. As of now, even for model species, the lncRNA entries in reference genomes are not exhaustively comprehensive. Hence, during analysis, there is often a requirement to assemble or predict new transcripts (those not included in the reference genome). Subsequently, these newly assembled transcripts need to be evaluated for their coding potential to procure the sequence and expression data of the newly predicted lncRNAs. lncRNA-seq can analyse the expression and differential information of mRNA, the expression and differential information of known lncRNAs (included in the reference genome) and newly predicted lncRNAs, and possible regulatory relationships between lncRNAs and mRNAs. The analyses that can be conducted in ordinary transcriptome sequencing can also be implemented in lncRNA-seq.
The data analysis and identification of lncRNAs are listed as follows:
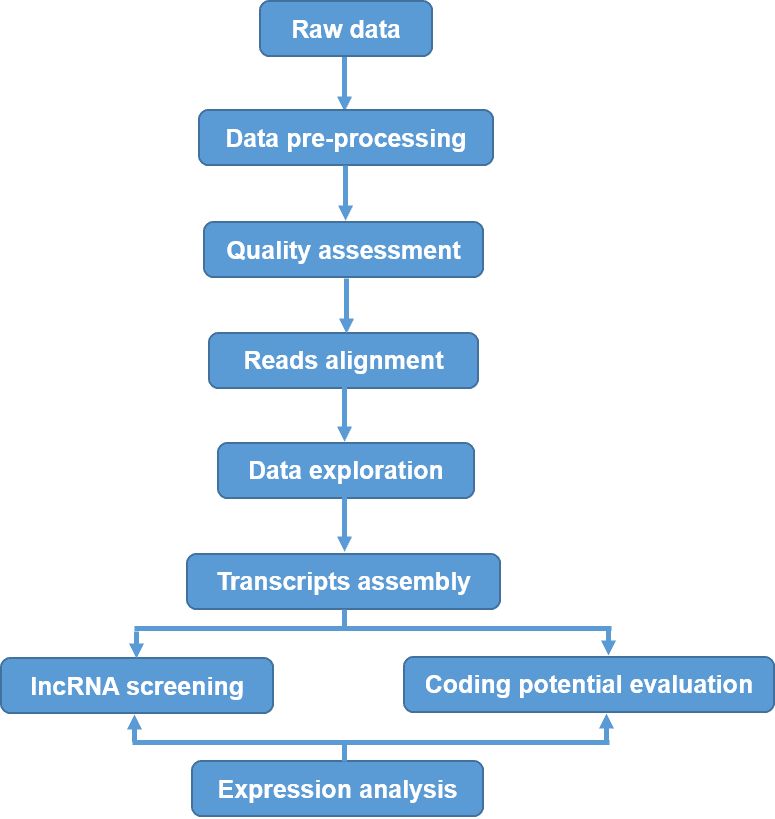 Figure 3. Overview of the data analysis and identification of lncRNAs.
Figure 3. Overview of the data analysis and identification of lncRNAs.
(1) Data pre-processing
- Quality assessment. After obtaining the raw data (fastq files), the quality of the original reads including sequencing error rate distribution and GC content distribution, is evaluated using FastQC v0.11.3.
- Data filtering. The original sequencing sequences contain low quality reads and adapter sequences. To ensure the quality of data analysis, raw reads must be filtered to get clean reads, and the subsequent analysis is based on clean reads. Data filtering mainly includes the removal of adapter sequences in the reads, the removal of reads with high proportion of N (N denotes the unascertained base information), and the removal of low-quality reads. This process is carried out using Cutadapt and Trimmomatic.
(2) Overall quality assessment of RNA-seq. It mainly includes inter-sample correlation assessment (Pearson correlation coefficient) and uniform distribution evaluation.
(3) Reads alignment to the Reference Genome. STAR aligner and Tophat 2 are often used for reads alignment. If the reference genome is properly selected and there is no contamination in the experiments, the results of the mapping (total mapped reads or fragments) would normally be higher than 70%.
(4) Data exploration. After the files are sorted, DESeq2 is used for data exploration. The output results can be used for cluster analysis and PCA (principal component analyses) analysis among RNA-seq samples, and the relationship among samples can be explored or the experimental design can be verified. The closer the sample clustering distance or PCA distance is, the more similar the sample is.
(5) Transcripts assembly. The transcripts are assembled with Cufflinks or Scripture software. Cufflinks uses the probability model to assemble and quantify the expression level of the isoform set as small as possible at the same time, to provide the maximum likelihood explanation of expression data at the mapping point, and to provide the chain information accurately with specific parameters for the chain specific library. Scripture, which is based on statistical segmentation model to distinguish between expression sites and experimental background noise, provides information about all isoforms with statistically significant expression at the mapping site, and is applicable to the assembly of long transcripts.
(6) Candidate lncRNA screening
- Basic screening. The basic screening consists of three steps: transcripts, whose length is greater than 200bp and the number of exons is greater than 2, are selected firstly; then, the coverage of each transcript is calculated by Cufflinks, and the transcripts with the minimum coverage of reads greater than 3 are selected; finally, non-lncRNAs are filtered out by comparison with known non-lncRNA, and the results of Cuffmerge are used for position screening (different class-code is selected for different kinds of lncRNA).
- Coding Potential Evaluation. The coding potential is the key factor to judge lncRNA. At present, the mainstream methods of encoding potential analysis include Coding Potential Calculator (CPC) analysis, Coding-Non-Coding Index (CNCI) analysis, PFAM protein domain analysis and PhyloCSF analysis.
(7) Expression analysis. It mainly includes expression level comparison, differential expression analysis, and differential expression lncRNA screening, lncRNA expression cluster analysis and tissue or phenotypic specific analysis. These analyses are usually carried out using DESeq or Cuffdiff.
(8) Advanced analysis
- LncRNA target gene prediction. The function of lncRNA is related to the adjacent coding protein gene. The protein coding genes adjacent to lncRNA are identified for functional enrichment analysis, and the main function of lncRNA could be predicted in lncRNATargets.
- Functional enrichment analysis of specific lncRNA. Specific lncRNA generally refers to the lncRNA with differential expression or tissue or phenotypic specific expression. The functional enrichment analysis of these specific lncRNAs by GO and KEEG can be performed respectively.
- Interaction analysis. LncRNA and mRNA can be related through the targeting relationship, and the mRNA can be related by protein, thus forming the interaction network of lncRNA-mRNA-protein. This interaction can be visualized by Cytoscape.
Using Wei et al.'s study titled "Long-chain noncoding RNA sequencing analysis reveals the molecular profiles of chemically induced mammary epithelial cells," we will explicate the data analysis of LncRNA sequencing.
Identification of lncRNA transcripts:
Coding potential analysis using various software tools such as CNCI, CPC, Pfam-scan, and CPAT. Noncoding transcripts identified by these tools were counted, and common and unique transcripts from each method were visualized using Venn diagrams.The intersection of predicted results was considered for subsequent analysis as the dataset for novel lncRNA.
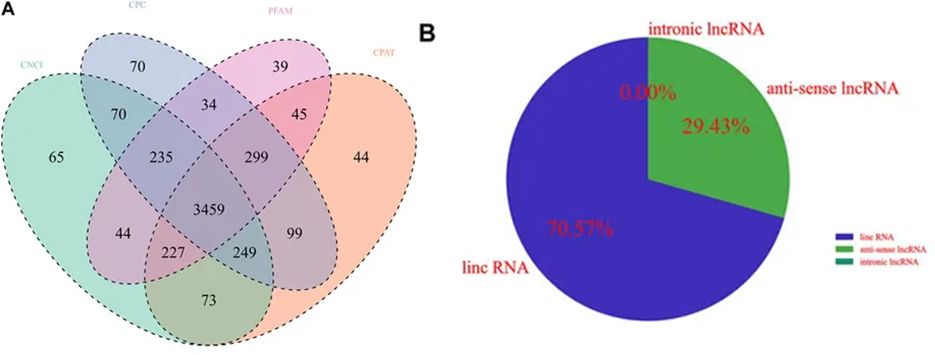 Figure 4. Identification of lncRNA transcripts.
Figure 4. Identification of lncRNA transcripts.
Structure and characterization of lncRNAs:
Through bioinformatics analysis, one can compare aspects of lncRNAs and messenger RNAs (mRNAs) such as their transcript length, the number of exons, and their respective expression levels. The results can reveal the concentrated area of lncRNA lengths, the quantity of exons in lncRNAs, as well as provide a comparative measure of global expression volumes between mRNAs and lncRNAs.
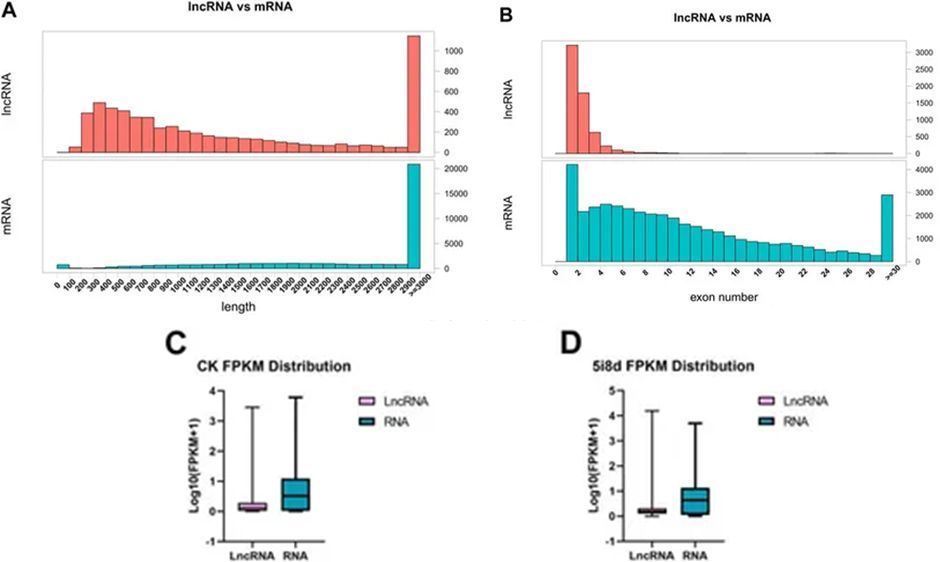 Figure 5. Comparative analysis of structural features of lncRNA and mRNA.
Figure 5. Comparative analysis of structural features of lncRNA and mRNA.
Differential expression analysis of lncRNAs:
DESeq was employed for differential expression analysis using criteria of |log2Fold Change|≥2 and p < 0.05. Identified significantly differentially expressed lncRNAs included both known and novel transcripts. Heatmaps were generated to visualize the expression patterns of differentially expressed lncRNAs between groups.
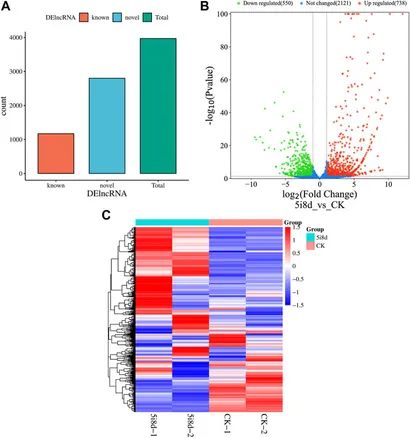 Figure 6. Differentially expressed lncRNA analysis.
Figure 6. Differentially expressed lncRNA analysis.
Differentially Expressed lncRNA-targeted mRNA Prediction:
Prediction of target genes of DE lncRNAs using both cis and trans target analysis methods. Identification of common target genes among multiple significantly different lncRNAs. Heatmap visualization of differently expressed target genes between experimental groups.
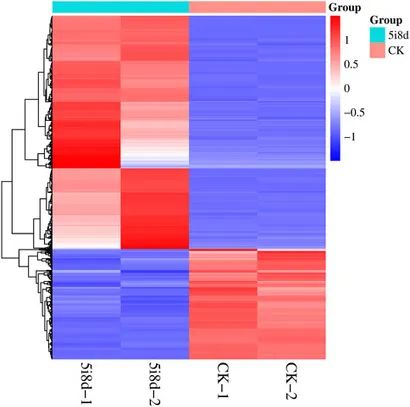 Figure 7. Heatmap of differentially expressed target genes.
Figure 7. Heatmap of differentially expressed target genes.
Validation of lncRNAs and Target Genes Expression by RT-PCR:
Six DE lncRNAs and four candidate target genes were selected for validation using RT-qPCR. Expression levels of selected lncRNAs and target genes were measured in goat fibroblasts and induced mammary epithelial cells. RT-qPCR results were consistent with the expression trends observed in transcriptome sequencing data, confirming the accuracy of sequencing results.
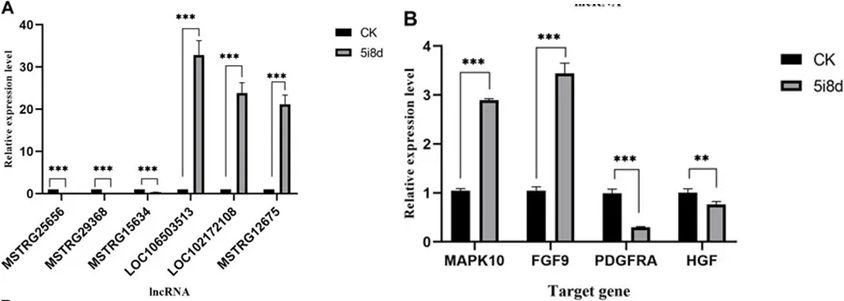 Figure 8. Comparison of the results of lncRNA-seq and RT-qPCR analysis of DE lncRNAs and target genes in CK group vs. 5i8d group.
Figure 8. Comparison of the results of lncRNA-seq and RT-qPCR analysis of DE lncRNAs and target genes in CK group vs. 5i8d group.
Functional Annotation Analysis of Target Genes: GO and KEGG:
Gene Ontology (GO) results are divided into three categories: Biological Process (BP), Cellular Component (CC), and Molecular Function (MF). Through the implementations of GO enrichment analyses, it was revealed that these target genes take dynamic roles in regulating myriad biological processes, including the modulation of metabolic processes, protein synthesis, receptor binding, and the regulation of tyrosine kinase activity. The enrichment analysis stemming from the Kyoto Encyclopedia of Genes and Genomes (KEGG) has provided additional insights, aiding the understanding of the biological pathways and signal transduction networks in which these target genes are implicated.
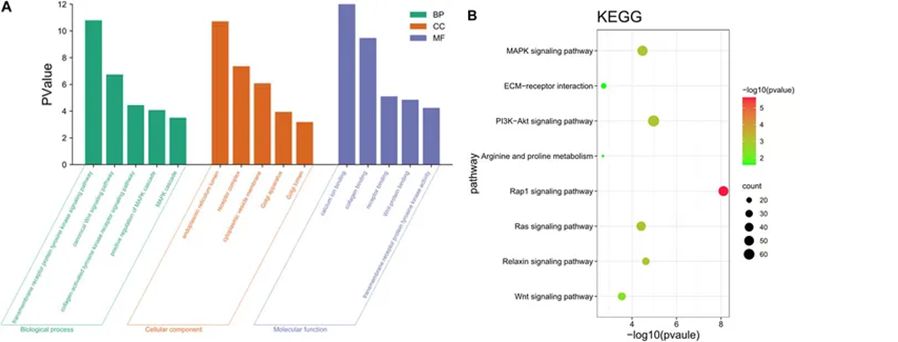 Figure 9. Functional annotation analysis of target genes.
Figure 9. Functional annotation analysis of target genes.
Interaction Network of lncRNA and mRNA:
Additionally, based on the recognized differential expression of lncRNA and mRNA genes, along with the predicted cis and trans target genes of lncRNA, one could construct a regulatory network analysis to depict the intricate interplay between differentially expressed lncRNAs and their respective target genes.
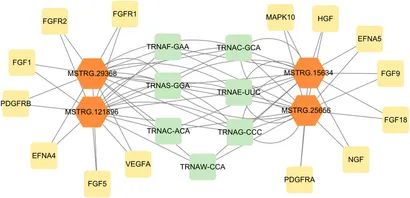 Figure 10. The regulatory network of differentially expressed lncRNAs and target genes.
Figure 10. The regulatory network of differentially expressed lncRNAs and target genes.
The Advantage of LncRNA Sequencing
Library Optimization: Employing ribosomal RNA depletion and strand-specific library construction methods, our approach preserves the integrity of both lncRNA and mRNA sequences, as well as their directional information.
Comprehensive Sequencing: Nearly all lncRNA and mRNA sequences in the samples were identified and analyzed, ensuring comprehensive coverage.
High Sensitivity and Resolution: Leveraging high-throughput sequencing technology, LncRNA sequencing enables the detection and quantification of low-expressed LncRNAs with remarkable sensitivity and resolution.
Species Diversity: We conducted identification and analysis of lncRNAs across a wide range of species, encompassing humans, animals, and plants, underscoring the diverse taxonomic scope of our investigation.
Dynamicity: LncRNA sequencing serves as a powerful tool for studying the dynamic expression patterns of lncRNAs, encompassing changes across different developmental stages, tissues, and physiological states. This elucidates the spatiotemporal regulatory mechanisms of lncRNAs in various biological processes.
Rigorous Analysis: Our approach involved meticulous curation of lncRNA databases for known lncRNA identification and the implementation of stringent filtering criteria to discover novel lncRNAs, ensuring the precision and reliability of our analysis.
Comprehensive Correlations: Embark on a thorough operation by associating lncRNA with mRNA, delving deeper into the regulatory networks governed by lncRNA.
Individual Variability: lncRNA sequencing can unveil differences in lncRNA expression between individuals, aiding the comprehension of variations in individual-specific expression of lncRNA and its role in diseases unique to certain individuals.
Starting Point for Functional Research: Discrepant lncRNA expressions can be sifted through lncRNA sequencing. This information serves as a prime pivot for research endeavors, further exploring their function and regulatory mechanics within biological processes.
Application of LncRNA Sequencing
The application of full-length lncRNA sequencing technology has connected cell types with their destiny, state, and functionality. This has accelerated developments within fields including developmental biology, oncology, neuroscience, immune-infection, and environmental toxicology.
Exploring the molecular mechanisms of lncRNAs in development and growth. For instance, Li et al. discovered a novel trans-acting lncRNA managed by the histone H3 on lysine 4 acetylation epigenetic mark of the ACTG1 gene. These findings build the theoretical foundation for future upstream and downstream functional analyses of lincRNAs in Brassicaceae. In addition, Yu et al. found that under high light intensity, the apple lncRNA MdLNC610 is involved in the accumulation of apple skin anthocyanins via activating ethylene biosynthesis. These cutting-edge findings demonstrate the pivotal roles played by lncRNAs in plant growth and development.
The construction of full-length lncRNA atlases across different species/tissues/diseases allows the determination of lncRNA expression patterns within phylogenetic lineages. For instance, Kyle Palos and colleagues constructed a Long intergenic noncoding RNAs (lincRNAs) database of four Brassicaceae species by assembling numerous lincRNA databases, as well as short-read and long-read RNA-seq technique. Not only were they able to confirm the conserved sequences of full-length lincRNAs, but they also established their functional elements, such as sORFs, structural regions, and miRNA interaction sequences, thereby laying the theoretical foundation for further analysis of lncRNA upstream and downstream functionalities in the Brassicaceae family.
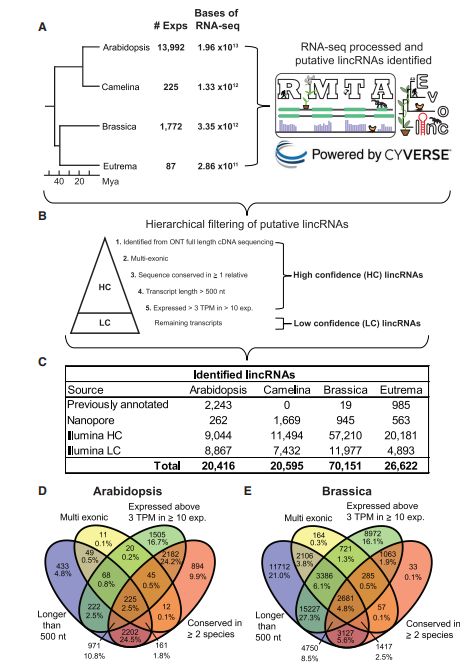 Figure 11. Identification and basic characteristics of lincRNAs in four cruciferous plants. (Palos et al., 2022)
Figure 11. Identification and basic characteristics of lincRNAs in four cruciferous plants. (Palos et al., 2022)
In the field of oncology, analyzing abnormal gene splicing in tumor cells and microenvironment samples and investigating the key lncRNA participants and regulators in the splicing process can help decipher the molecular mechanisms of lncRNA in tumor progression. This could also aid in the development of early tumor diagnostic molecular markers. For instance, Lan et al. have found that lncRNA SNHG6 can induce hnRNPA1 to splice mRNA PKM specifically, thereby increasing the PKM2/PKM1 ratio, enhancing aerobic glycolysis in colorectal cancer cells, and thus promoting carcinogenesis.
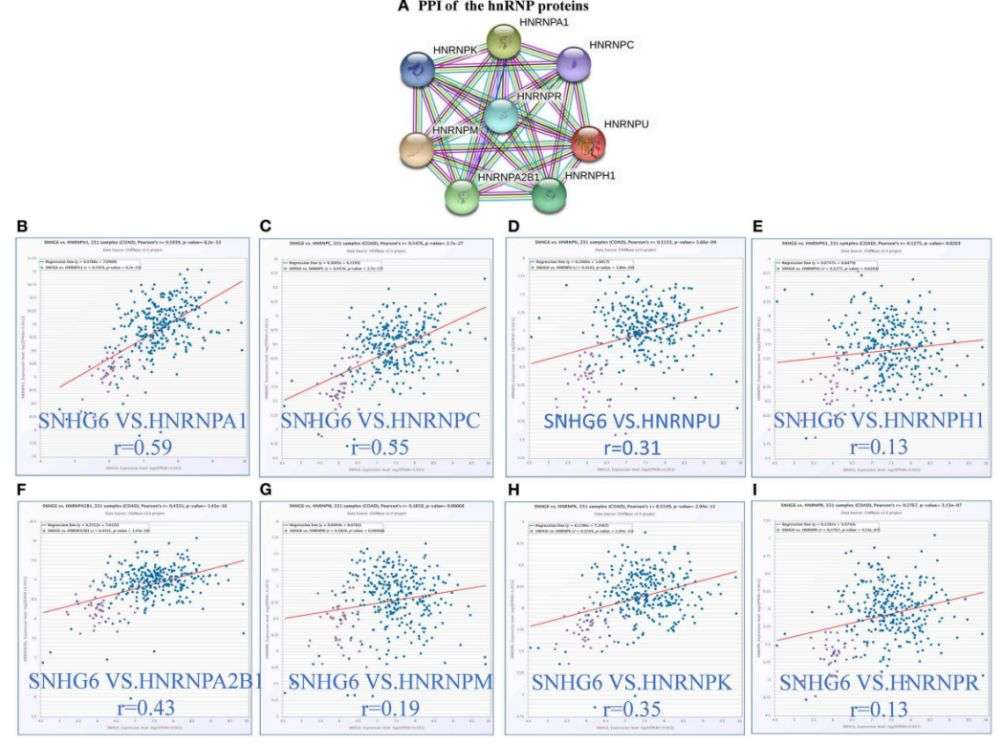 Figure 12. The expression of hnRNP proteins is positively correlated with the expression of SNHG6 in colorectal cancer. (Lan et al., 2020)
Figure 12. The expression of hnRNP proteins is positively correlated with the expression of SNHG6 in colorectal cancer. (Lan et al., 2020)
The study of host immune cell responses, revealing the functional regulation of long non-coding RNAs (lncRNAs) involved in host immune responses, is of paramount importance. Recent research findings demonstrate that the transcription factor FUBP3 can serve as an RNA-binding protein, interacting synergistically with lncRNA EST12 to suppress the expression of cytokines such as Interleukin (IL)-1β and IL-6, thereby promoting the proliferation of Mycobacterium tuberculosis (MTB). It has been observed that infection with the standard MTB strain H37Rv leads to the downregulation of lncRNA GAS5 expression in macrophages, and by targeting miR-18a-5p, there is a consequential enhancement of macrophage vitality. In parallel, the expression levels of lncRNAs NORAD and SNHG16 increase after infection. These lncRNAs negatively regulate their respective target genes miR-618 and miR-140-5p, achieving control over macrophage proliferation and inflammation responses, in turn influencing the outcome of MTB infection.
The research delves into the alterations in the expression of long non-coding RNAs (lncRNAs) at the cellular level under the stress of various types of environmental pollutants. These pollutants include polycyclic aromatic hydrocarbons, polychlorinated biphenyls, polybrominated diphenyl ethers, benzene, bisphenol compounds, per- and polyfluorinated alkyl substances, pharmaceuticals and personal care products, pesticides, heavy metals, nanomaterials, and PM2.5 particles. The goal is to unravel the issues at the intersection of environmental toxicology and reveal the molecular mechanisms by which lncRNAs contribute to the toxicity effects of environmental contaminants. Cao et al. selected several well-studied lncRNAs including Birc6-AS2, Mettl3, Malat1, Stedlb1a, and Oip5-AS as reference targets. They exposed zebrafish embryos to solutions of mercury chloride, methylmercury, lead chloride, cadmium chloride, and chromic acid. They found differential expression of lncRNA Malat1 under mercury chloride and methylmercury exposure. Concurrently, the mercury-exposed group of zebrafish embryos exhibited noticeable neurobehavioral disorders, indicating that the mercury-specific induction of lncRNA is involved in mercury's neurotoxic effects.
If you are interested in our lncRNA sequencing service, please feel free to contact our scientists. In addition to this, we provide a package of transcriptomics sequencing services involving RNA-seq, small RNA sequencing, circRNA sequencing, degradome sequencing, and bacterial RNA sequencing.
References:
- Arrigoni, A., Ranzani, V., Rossetti, G., Panzeri, I., Abrignani, S., Bonnal, R. J. P., et al. (2016). Analysis RNA-seq and noncoding RNA. Polycomb Group Proteins. Springer New York.
- Anders, S. and Huber, W. (2010) 'Differential expression analysis for sequence count data'. Genome Biology. 11: R106.
- Guo, X., Gao, L., Wang, Y., Chiu, D. K., Wang, T. and Deng, Y. (2015). Advances in long noncoding RNAs: identification, structure prediction and function annotation. Briefings in Functional Genomics, 15(1), 38-46.
- Guttman, M., Garber, M., Levin, J. Z., Donaghey, J., Robinson, J., Adiconis, X., et al. (2010). Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nature Biotechnology, 28(5): 503-510.
- Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocol, 7(3): 562-578.
- Wei M, Tang W, Lv D, et al. Long-chain noncoding RNA sequencing analysis reveals the molecular profiles of chemically induced mammary epithelial cells. Frontiers in Genetics, 2023, 14: 1189487.
- Li N, Zhou Y, Cai J, et al. A novel trans-acting lncRNA of ACTG1 that induces the remodeling of ovarian follicles. International Journal of Biological Macromolecules, 2023, 242: 125170.
- Palos K, Nelson Dittrich A C, Yu L, et al. Identification and functional annotation of long intergenic non-coding RNAs in Brassicaceae. The Plant Cell, 2022, 34(9): 3233-3260.
- Yu J, Qiu K, Sun W, et al. A long noncoding RNA functions in high-light-induced anthocyanin accumulation in apple by activating ethylene synthesis. Plant Physiology, 2022, 189(1): 66-83.
- Lan Z, Yao X, Sun K, et al. The interaction between lncRNA SNHG6 and hnRNPA1 contributes to the growth of colorectal cancer by enhancing aerobic glycolysis through the regulation of alternative splicing of PKM. Frontiers in oncology, 2020, 10: 363.
- Cao M, Song F, Yang X, et al. Identification of potential long noncoding RNA biomarker of mercury compounds in zebrafish embryos. Chemical research in toxicology, 2019, 32(5): 878-886.


