
 Sample Submission Guidelines
Sample Submission Guidelines
RNA-Seq for Differential Gene Expression Analysis: Introduction, Protocol, and Bioinformatics
Introduction of RNA-Seq for Differential Gene Expression
Generating a comprehensive expression profile is critical when studying normal biology and disease processes. The transcriptome is a cell's or population's complete set of transcripts, and transcriptome analysis reveals the identity and quantity of all RNA molecules. The correlation of transcriptomes across developmental stages, disease states compared to normal cells, or specific experimental stimuli compared to physiologic conditions is an essential application of RNA-seq. This sort of analysis necessitates the identification of genes and their isoforms, as well as a precise estimation of their abundance when comparing two or more samples. It's crucial for deciphering the genome's functional elements and determining the molecular makeup, which can lead to new insights into the biological mechanisms of development and disease. Cuffdiff, DESeq, DESeq2, EdgeR, PoissonSeq, Limma voom, and MISO are some of the most used tools for differential gene expression.
Services you may interested in
Protocol and Bioinformatics in RNA-Seq for Differential Gene Expression Analysis
Following the step of preprocessing RNA-seq reads, DGE analysis is used to determine how the transcript levels differ between samples. Since the microarray era, numerous statistical techniques have been established that use read coverage to assess transcript abundance. The RPKM (reads per kilobase per million mapped reads) technique is widely used to account for expression and normalized read counts in relation to the total number of mapped reads and gene length. However, in addition to read coverage, other factors such as sequencing depth, gene length, and isoform abundance influence the approximated transcript abundance. It has been critiqued because the RPKM method treats all RNA-seq reads almost equally, for example, without regard for isoforms. RNA-Seq by Expectation-Maximization (RSEM) is a newly developed software tool that provides accurate gene and isoform expression levels for species without a reference genome assembly.
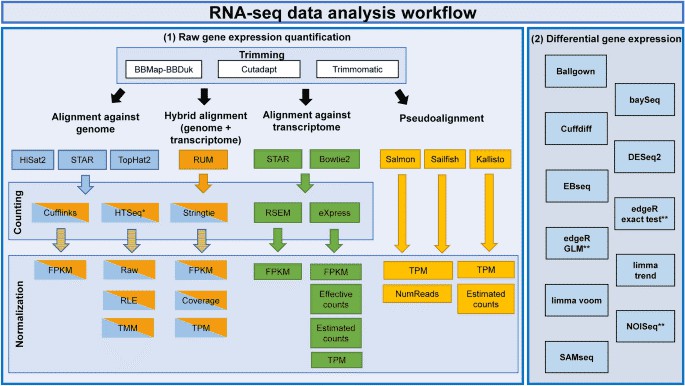 Figure 1. RNA-seq analysis workflow for gene expression. (Corchete, 2020)
Figure 1. RNA-seq analysis workflow for gene expression. (Corchete, 2020)
To date, most differential gene expression analysis algorithms use simple count-based probability distributions followed by Fisher's exact test without taking into account biological variability between samples. While RNA-seq data has very low technical variability when compared to microarray data, biological variability can be substantially lowered by evaluating multiple replicates using permutation-derived methods. For biological variability assessment, serial analysis of gene expression has been developed, in which larger-scale datasets are used to approximate an additional dispersion parameter based on an extended Poisson distribution, enabling extensive molecular characterization capability.
However, a large number of replicas may be too expensive for most applications, so many established techniques have surmounted the problem by modeling biological variability and measuring significance with a small number of samples, using pairwise or multiple group comparisons. Several programs provide well-designed solutions for this purpose, and they have been used in numerous biomedical and clinical studies. Cuffdiff from the Cufflinks package, DESeq, DESeq2, and EdgeR are examples of these programs. Because RNA-seq read counts are highly skewed integer numbers ranging from zero to millions, a variety of transformation algorithms have been used to fit the counts to statistical distribution models for differential expression detection. For RNA-seq counts, approaches developed for microarray data analysis based on continuous distribution have been improved. The voom function in the limma package is an excellent example of how to transform count data into Gaussian distributed data so that statistical significance can be tested. A comprehensive comparison of the performance of several DGE packages was recently published. However, there is no one-size-fits-all strategy that we are aware of.
The Differential Gene Expression (DGE) tool conducts statistical tests based on the quantification of expressed genes derived from the computational analysis of raw RNA-seq reads, such as mapping and assembly, to determine which genes exhibit statistically significant differences. It also provides information related to the expression level of each gene and the magnitude of pairwise differences. DGE analysis can offer substantial insights into the genetic mechanisms underlying phenotypic differences in organisms, including plant growth patterns, detection of tumor origins, and microbial community studies.
Algorithms for Read Mapping
The initial computational step in the RNA sequencing data analysis pipeline involves read mapping, a fundamental process aimed at aligning sequence reads to the reference genome or transcriptome by identifying genomic regions that match the read sequences. A variety of mapping tools have been developed to facilitate this process. In all instances, the mapping process commences with the construction of an index for the reference genome or read set, enabling rapid retrieval of potential locations in the reference sequence where reads are most likely to align. Subsequently, slower yet more sensitive algorithms are employed to align reads within these candidate regions.
Digital Measurement of Gene Expression
Upon mapping, each sequenced read aligned to a coding unit (such as an exon, transcript, or gene) is employed in counting, thereby estimating its expression level. The most prevalent method for counting involves tallying the number of reads overlapping with gene exons. In the analytical workflow, quantification of gene expression in RNA sequencing data is typically achieved through two computation steps: aligning reads to a reference genome or transcriptome and subsequently estimating the abundance of genes and isoforms based on the aligned reads. Regrettably, reads generated by the most commonly used RNA sequencing technologies are often much shorter than the transcripts sampled from which they originate. Consequently, in scenarios where transcripts with similar sequences exist, it is not always feasible to uniquely assign short reads to specific genes.
Quantification Biases and Normalization
Numerous studies emphasize the necessity for meticulous data normalization prior to assessing differential gene expression in order to correct for various sources of bias. The first bias to consider is the "sequencing depth" of the sample, defined as the total number of reads sequenced or mapped. RNA sequencing counts also exhibit biases related to gene length: the expected number of reads mapping to a gene is proportional to the abundance and length of the isoforms transcribed from that gene.
Data Mining Analysis
In recent years, research in the field of RNA sequencing has flourished, leading to the emergence of various differential gene expression detection tools. The simplest methods for detecting differential gene expression rely on statistical tests to determine which genes exhibit statistically significant changes in expression under different conditions. Nonparametric methods could theoretically be employed. However, due to the limited number of replicates typically available in RNA sequencing experiments, nonparametric methods often lack sufficient detection power, making parametric methods more popular. Given a specific count of RNA sequences, all tools for analyzing differential gene expression include two primary steps: estimating model parameters based on the data and detecting differentially expressed genes using statistical tests.
Conclusion
RNA-Seq is a cutting-edge technology revolutionizing the analysis of differential gene expression, providing an unparalleled insight into the transcriptomic landscape. This method empowers researchers to pinpoint and measure RNA molecules with remarkable accuracy, unravelling the intricate mechanisms of gene regulation in both physiological and pathological conditions. The adaptability of RNA-Seq transcends disciplinary boundaries, spanning from fundamental biological research to clinical investigations, establishing itself as an indispensable asset in contemporary molecular biology. Advancements in sequencing methodologies promise to enhance the efficacy and accessibility of RNA-Seq further. The future landscape may feature refined single-cell RNA-Seq techniques, long-read sequencing for improved transcriptome reconstruction, and integrated multi-omics strategies amalgamating RNA-Seq data with diverse molecular datasets to offer a comprehensive understanding of cellular processes.
If you want to learn more about the analysis of differential gene expression and the data analysis process of RNA sequencing, you can refer to our articles "What is Differential Gene Expression Analysis?" and "Bioinformatics Workflow of RNA-Seq".
References:
- Wang T, Li B, Nelson CE, Nabavi S. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC bioinformatics. 2019 Dec;20(1).
- Lamarre S, Frasse P, Zouine M, et al. Optimization of an RNA-Seq differential gene expression analysis depending on biological replicate number and library size. Frontiers in plant science. 2018 Feb 14;9.
- Costa-Silva J, Domingues D, Lopes FM. RNA-Seq differential expression analysis: An extended review and a software tool. PloS one. 2017 Dec 21;12(12).
- Corchete LA, Rojas EA, Alonso-López D, et al. Systematic comparison and assessment of RNA-seq procedures for gene expression quantitative analysis. Scientific reports. 2020 Nov 12;10(1).
- McDermaid A, Monier B, Zhao J, et al. Interpretation of differential gene expression results of RNA-seq data: review and integration. Briefings in bioinformatics, 2019, 20(6): 2044-2054.
- Finotello F, Di Camillo B. Measuring differential gene expression with RNA-seq: challenges and strategies for data analysis. Briefings in functional genomics, 2015, 14(2): 130-142.


