
 Sample Submission Guidelines
Sample Submission Guidelines
Overview of DNA Sequencing
Genomic sequencing technology, also referred to as DNA sequencing technology, entails the acquisition of the nucleotide arrangement of a targeted DNA segment. In the realm of scientific research, obtaining the sequence of a target DNA fragment serves as the foundational step for subsequent molecular biology investigations and genetic modifications. As the landscape of genomics, particularly propelled by advancements such as Genome-Wide Association Studies (GWAS), continues to mature, the anticipation of disease risk and early diagnosis through genomic approaches has emerged as a pivotal trajectory in clinical medicine. Furthermore, precision medicine, heralded as a key avenue in the future of clinical medicine, underscores the undeniable significance of genomic sequencing technology within its paradigm. Each evolution in sequencing technology stands as a monumental impetus for diverse fields, including genomic research, disease therapeutics, and pharmaceutical development. These transformative strides continually contribute to the acceleration and enrichment of our understanding and capabilities in genomics, ultimately shaping the landscape of medical research and practice.
Development of Sequencing Technology
The evolution of DNA sequencing technology has been remarkable since its inception with the first-generation Sanger method. Based on differing principles, the progress of sequencing technology can be segmented into three stages: first-generation Sanger sequencing, second-generation high-throughput sequencing, and third-generation single-molecule/nanopore sequencing. Each generation has its own strengths and weaknesses; hence, their applications vary accordingly. Currently, the sequencing market is dominated by Next-Generation Sequencing (NGS), but it also accommodates the co-existence of Long-read sequencing (Long-read sequencing) technologies.
Service you may interested in
Sanger Sequencing
The first-generation sequencing technologies encompassed the Sanger double-deoxy nucleotide terminal termination method, and the chemical degradation method innovated by Alan Maxam and Walter Gilbert. This groundbreaking technology facilitated automated sequencing of nucleic acid molecules. As of 1977, first-generation sequencing could achieve a sequencing length of 1,000 base pairs (bp) with a high accuracy rate of 99.99%, providing an advanced tool for molecular biology research. For a considerable period, this method was highly esteemed for its long reads, high sensitivity, and accuracy, playing a crucial role in the early Human Genome Project. However, Sanger sequencing has its limitations; although the technique provides longer reads compared to subsequent second-generation sequencing, each sequencing run can only yield a sequence of about 700-1000 base pairs in length, as the length of each read is the limit of its single-run sequencing capacity. Given that a separate primer design is requisite for each sequencing, when confronted with the widespread demand for genome-scale sequencing in modern research and clinical applications, the relatively lower throughput, time-consuming process, and higher costs of Sanger sequencing have limited its prospects for large scale applications.
Gradually, the dominance of first-generation sequencing was replaced by the emerging second-generation sequencing (Next Generation Sequencing or NGS). However, the first-generation sequencing technology still possesses certain advantages, such as high accuracy, longer reads, and eliminating the need for sequence assembly. Therefore, it maintains significance in some research fields. For instance, it plays an instrumental role in areas such as plasmid sequencing, mutation site validation, small bacterial genome sequencing, bacterial artificial chromosome-end sequencing, and repetitive sequence sequencing.
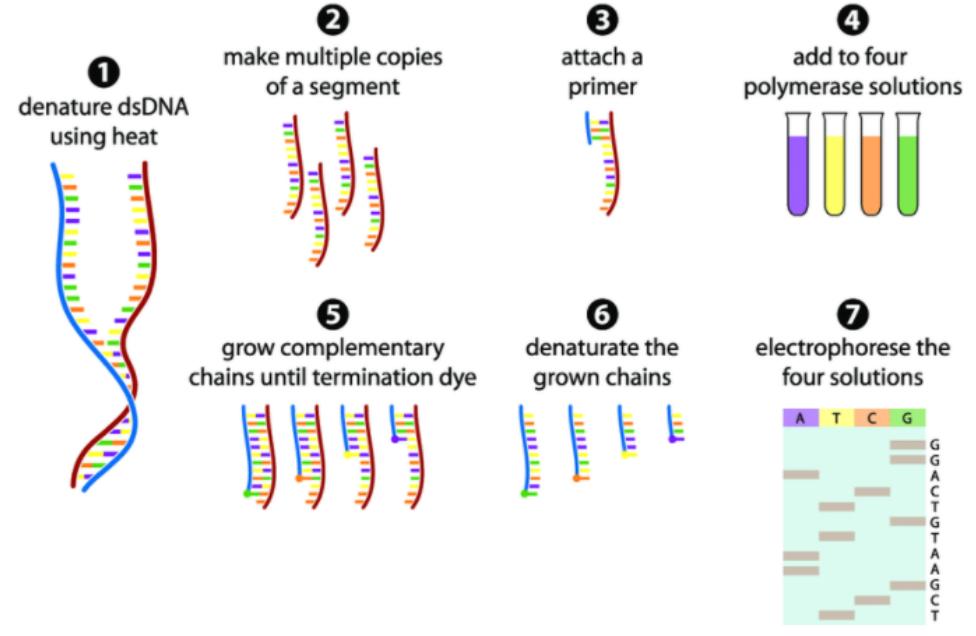 Figure 1 Principle of Sanger sequencing (Michel G Gauthier 2007)
Figure 1 Principle of Sanger sequencing (Michel G Gauthier 2007)
Next-Generation Sequencing
High-Throughput Sequencing (HTS), also known as Next-Generation Sequencing (NGS) or second-generation sequencing, mainly incorporates the 454 technique from Roche, the HiSeq technique from Illumina, the SoLiD technique from ABI, and the Ion Torrent technique from Thermo Fisher. Both the 454 and SoLiD techniques have been discontinued owing to their complex nature, high cost, and inconsistent sequencing stability. The NGS technologies widely used in the current market primarily derive from Illumina and Thermo Fisher; companies like BGI are also promoting NGS products.
In 2008, Applied Biosystems (ABI) and Invitrogen merged to create Life Technologies, which subsequently launched the sequencers Ion Torrent and Ion Proton in 2010 and 2012, respectively. By 2014, Life Technologies was acquired by Thermo Fisher Scientific for a substantial $13.6 billion. As a result of this transaction, Thermo Fisher has mass-manufactured and vigorously promoted the core products of Life Technologies, specifically their clinical-grade sequencers.
Meanwhile, in 2014, Life Technologies was acquired by the Thermo Fisher Corporation for a substantial sum of $13.6 billion. This significant acquisition transitioned the function of the foremost products of Life Technologies. Originally used for research purposes, these sequencing devices were transformed into clinical grade sequencers, mass-produced and seamlessly integrated into Thermo Fisher's repertoire. The decision made by Thermo Fisher to promote these products was a testament to their potential in transforming the landscape of clinical genomics.
The advent of 454 sequencing technology has served to mitigate the shortcomings inherent in the first-generation sequencing approaches. It exhibits distinctive traits such as high throughput (with an average output reaching up to 0.5G), cost-efficiency, and time-saving processes. However, when sequencing sequences abundant in repetitive nucleotide structures or sequences, the Sanger sequencing methodology proves more appropriate.
Solexa sequencing technology rose to prominence during a period characterized by rapid advancements in the field of gene chips and has since found extensive applications in the sequencing of animals, plants, and microbes. Similar to 454 sequencing technology, Solexa operates on the principle of "sequencing by synthesis", boasting advantages such as negating the need for probe synthesis and an understanding of the model organism's genome sequence, thus simplifying the proceedings.
Moreover, this technology employs integrated chips during the preparation stage to connect DNA molecules with specific primers, facilitating their specific binding to the chip. This results in "monoclonal" DNA and confers traits such as high throughput (with an average output reaching up to 30G), a high level of sensitivity, and enhanced accuracy upon the technique.
The SOLiD (Supported Oligo Ligation Detection) sequencing method represents a divergent technological approach in contrast to the previously mentioned strategies. Its underlying principle revolves around ligation rather than PCR amplification. The targeted DNA sequence is derived through multiple rounds of sequencing following the binding of the target DNA with octa-base fluorescent probes. By eliminating the "synthesis" step, this method effectively minimizes mismatch phenomena during base pairing, thereby laying a foundation for improved accuracy.
In comparison to Sanger sequencing, Next-Generation Sequencing (NGS) has sharply broken through in terms of throughput, overcoming the previous generation's modulus operandi which restrained the sequencing to only one piece of DNA at a time. A single run of NGS may concurrently reveal hundreds of thousands to millions of nucleotide sequences, thereby dramatically diminishing the time required for genome-scale sequencing. Moreover, the widespread application of NGS technology is greatly attributable to its reduced cost.
Service you may interested in
Long-read sequencing
The Long-read sequencing techniques, known as PacBio's Single Molecule Real-time (SMRT) technology and Oxford Nanopore Technologies' Nanopore technology, address deficiencies associated with Next-Generation Sequencing (NGS). Despite the revolution in throughput offered by NGS, its inherent limitations regarding short read length create obstacles during the sequencing and assembly of highly repetitive sequence regions. Furthermore, obtaining accurate gene sequence information from NGS requires not only high sequencing coverage but also precise sequence assembly techniques.
Long-read sequencing technologies offer a solution to these limitations, providing sequencing results for individual long sequences de novo. These technologies, also known as single-molecule sequencing techniques, accomplish this feat by performing sequencing on single long sequences, bypassing the need for PCR amplification of the target molecule. The result is that these methods directly yield nucleic acid sequence information spanning tens of thousands of base pairs while ensuring a high-throughput.
In October 2015, Pacific BioSciences (PacBio) introduced the Sequel system, a third-generation sequencer developed around their Single Molecule Real-Time (SMRT) technology. The Sequel is characterized by a metal chip endowed with numerous zero-mode waveguides (ZMWs)—actually circular apertures with a diameter of merely 100nm—containing DNA polymerase, the DNA sample to be sequenced, and fluorescently tagged deoxyribonucleotide triphosphates (dNTPs). The ZMWs channels focus the relevant light intently into a minute detection zone at the bottom which is then capable of registering the distinct fluorescence of each dNTP type, hence determining the base sequence of the nucleic acid.
The efficacy of the SMRT technology hinges on three key components: 1) DNA polymerase, where the length of the sequenced read principally hinges on the enzymatic activity of the polymerase. As this is susceptible to damage from the laser used in detection, it is a major consideration. 2) The labelling of the dNTPs with fluorescent entities at the 3' phosphate end. As the DNA synthesis progresses, the breaking of the 3' phosphodiester bond releases the tagged entities used for detecting, thereby minimizing steric hindrance and favoring uninterrupted DNA synthesis. This ensures the extension of the read's length. 3) The distinguishing function of the ZMW's apertures, which prove useful in discerning the reaction signals within the noise of strong surrounding fluorescence. Its structural strength is emphasized by the limited energy range due to laser light that is unable to penetrate the tiny orifices to reach the upper solution area. This ensures that fluorescent signals originate solely from the tiny reaction zone, leaving monomers outside the pore in darkness—effectually minimizing background fluorescence.
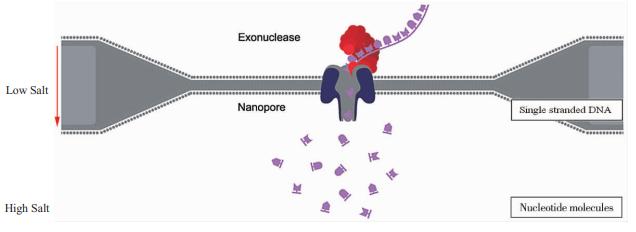 Nanopore sequencing technology (Image source Nanopore)
Nanopore sequencing technology (Image source Nanopore)
Simultaneously, Oxford Nanopore Technologies launched their third-generation sequencer, the MinION, hinging on Nanopore technology. The operational models include a special lipid bilayer with electrodes on one side, poised atop a nanopore. This bilayer contains various nanopores made up of alpha-hemolysin proteins, each accommodating a nucleic acid exonuclease. The DNA template moving into the nanopore binds with the exonuclease, which coordinates to successively sever the DNA bases transiting through the nanopore. When each base passes through, it engenders a unique current blockade. Collectively, the detection and interpretation of these minute current variations enable the determination of the related base type, resulting in the elucidation of the entire DNA molecule's sequence.
Table 1 Comparison of the advantages and disadvantages of next-generation sequencing and Long-read sequencing
| Next-Generation Sequencing | Long-read sequencing | |
| Advantages | - Capable of generating multiple reads for the same region with high sequencing depth, achieving high precision. | - Longer reads without the need for template references, reducing bioinformatics assembly costs, saving memory, and computation time. Can be used to determine highly repetitive sequences, improving sequencing coverage. |
| - Mature sample processing procedures with standardized DNA sample processing and reagents. | - Sequences directly from the original DNA sample, avoiding errors introduced by PCR amplification. | |
| - Sequencing costs have significantly decreased, ranging from $0.05-$0.15 per Mb. | - Expanded the application of sequencing technology, e.g., direct sequencing of RNA sequences, significantly reducing system errors introduced by reverse transcription. Direct sequencing of methylated DNA sequences, distinguishing between normal and methylated C bases by measuring DNA polymerase pausing times. | |
| - Highly mature bioinformatics software for rapid data processing, quality control, and assembly. | - Applicable to ctDNA and single-cell sequencing: High sensitivity of 3rd Gen sequencing allows monitoring of ctDNA at levels below 1 ng. At the single-cell level, direct sequencing of original DNA allows in situ sequencing after cell lysis. | |
| Disadvantages | - Shorter read lengths, time-consuming assembly, and lower coverage of repetitive and palindromic sequence regions. | - Higher error rates in single-read lengths, requiring repeated sequencing for error correction, significantly increasing sequencing costs. |
| - Introduction of systematic errors during synthesis and sequencing using PCR technology. | - High dependence on the activity of biological enzymes, with sequencing conditions directly affecting sequencing efficiency and accuracy. | |
| - Dependence on upstream processes such as reverse transcription and bisulfite treatment for non-DNA genome sequencing (e.g., RNA sequencing, DNA methylation sequencing), introducing a higher risk of errors. | - Higher cost, ranging from $0.33-$1.00 per 1 Mb. | |
| - Lower sensitivity for low-abundance DNA samples and challenging assembly without pre-existing reference templates. | - Insufficient variety in bioinformatics analysis software and limited data accumulation. |
Recent years have witnessed the robust adoption of Long-read sequencing technologies in genomic analyses. These methodologies have significantly propelled the progress of genomic exploration and decryption, realizing substantial advancements. Long-read sequencing, direct sequencing of individual DNA molecules, brings greater read lengths and enables high sensitivity, template-free assembly, and direct sequencing of DNA methylomes and RNA. However, limitations exist. Precise operational controls are required, costs are substantial, mature bioinformatics processing software is scarce, and error rates are high. Especially in cost and precision-demanding contexts, such as precision medicine and early cancer screening, the practical application of these technologies remains constrained.
Contrarily, while Next-Generation Sequencing (NGS) has drawbacks, including dependency on existing templates, short read lengths, and sequence bias, its mature system, high-throughput capacity, high sensitivity, high resolution, and low cost make it more accessible and widely adopted throughout various laboratories. Indeed, the relationship between Long-read sequencing and NGS is not characterized by competition or substitution, but rather, these technologies act as complementary assets.
Taking the sequencing of microbial resistance groups, for example, most resistance genes are located on plasmids, many of which are not successfully sequenced in preliminary research. Sole reliance on NGS cannot accurately obtain the plasmid sequences containing resistance genes. At this junction, Long-read sequencing can be leveraged to directly sequence the plasmid's single-molecule DNA. However, due to the low accuracy and precision of Long-read sequencing, upon obtaining the plasmid sequence, it is often necessary to use NGS to generate a large number of high-precision short reads for comparison, leading to a more accurate plasmid sequence.
As the cost of Long-read sequencing continues to decrease, more scenarios will require the combination of NGS and Long-read sequencing technologies to obtain precise unknown sequences in future clinical applications. These two technologies can complement each other, mutually enhancing their respective application values.
Service you may interested in
NGS Technology Principles and Processes
In 2005, the Genome Sequencer 20 (GS 20) system was introduced by the 454 company, pioneering the era of "Sequencing by synthesis" (SBS). Today, SBS methodology is foundational to Thermo Fisher's Ion Torrent and Illumina's next-generation sequencing (NGS) platforms.
The essence of SBS technology is common across platforms and mainly consists of three key steps: 1) Library preparation, which involves fragmenting long-chained DNA samples into short segments to create a DNA library. During fragmentation, adapters are attached to both ends of the short segments to facilitate subsequent fragment fixation, using the adaptors as primer binding sites for amplification processes; 2) Loading, in which the DNA library is allocated onto a sequencing medium and undergoes amplification. The objective of this step is to multiply the number of these short segments, generating a sufficient quantity of identical short fragments to amplify the signals in the subsequent sequencing phase. This allows the detection of sequence information; 3) Sequencing, where the amplified double-stranded DNA undergoes denaturation to form single-stranded DNA, which then serves as a template. The integration of single nucleotides during each round of synthesis is monitored and sequence information is discerned from the recording of signals (e.g., fluorescence, pH changes).
Differences between SBS NGS products offered by different companies emerge on aspects such as sample processing, signal detection, and fragment fixation.
The standard procedures for Next-Generation Sequencing technologies include the following:
For the 1.3.1454 sequencing technology, the process begins with fragmentation of the sample, followed by the selection of fragments between 500 and 800 base pairs (bp) in length. Adapters are then added to both ends of these fragments. The adapter-ligated DNA fragments are combined with magnetic beads, encapsulated in a water-in-oil system, and partnered with a reaction plate. The system is then incorporated into a PTP plate where the four base types are sequentially added for sequencing. Lastly, the signals are monitored and sequence data collected.
Solexa sequencing technology similarly commences with sample fragmentation. Subsequently, a library is established and amplified on a chip embedded with primers to produce single clones. The system then introduces four differently labeled dNTPs along with modified DNA polymerase. Collected fluorescent signals are recorded to extract sequence data.
The Solid sequencing technology begins by fragmenting the sample and attaching adapters to both ends of these fragments. This is followed by PCR amplification and microparticle enrichment. Magnetic beads are then settled in, allowing hybridization between the adapters and specific adapter sequences on the template. Four differently colored fluorescent dyed dual-base probes are connected with the sample. The final step involves detecting fluorescent signals and gathering sequence data.
Building upon the above descriptions, we have juxtaposed the features of three second-generation sequencing technologies alongside Sanger sequencing technology (table 1). A horizontal comparison of the second-generation sequencing technologies was also conducted (table 2). It can be inferred that compared to the first-generation Sanger sequencing, all second-generation sequencing technologies demonstrate high-throughput, high accuracy, and cost-efficiency.
Table 2: Comparison of Sequencing Technologies
| Technology | Principle | Sequencing Approach | Read Length | Accuracy | Error Types | Cost ($/Mb) | Throughput | Platform |
| 454 | Pyrosequencing | Simultaneous synthesis and sequencing | 500 bp | 99% | Insertions, deletions | Low | 250-400 Mb | Bead |
| Solexa | Fluorescence | Simultaneous synthesis and sequencing | 100 bp | 98.99% | Substitutions | Low | 3-6 GB | Glass |
| Solid | Fluorescence | Ligation-based sequencing | 50 bp | 99.94% | Substitutions | Much low | 30 GB | Glass |
| Sanger | Radioactive | Simultaneous synthesis and sequencing | 1000 bp | 99.99% | Substitutions | High | 1 Mb | PAGE |
Table 3: Comparison of Three Next-Generation Sequencing Technologies
| Sequencing Technology | Advantages | Disadvantages |
| Roche/454 | Long read lengths | Low throughput, high cost |
| Solexa | High throughput, high accuracy, low cost | Short read lengths, complex operation |
| Solid | High throughput, high accuracy, low cost | Short read lengths, complex operation |
Data Quality Control for NGS
The sequencing process in Synthesis-by-Sequencing (SBS) involves a notable problem: as the synthesized chain elongates, the efficiency of the DNA polymerase diminishes, concurrently reducing its specificity. This results in a gradual increment in the base synthesis error rate as sequencing progresses, accentuating the importance of quality control (QC) in Next-Generation Sequencing (NGS) data for subsequent analyses. This also becomes a crucial identifier when evaluating the data consistency and quality of sequencing instruments.
Within the QC framework for NGS outputs, the parameters of paramount importance include:
1) Q20, Q30 and Q40: In high throughput sequencing of genes, each base is allocated a quality value (Q), the higher the Q value, the lesser the chances of an erroneous base measurement, computed by the formula Q = -10logP. Q20 and Q30 represent the proportion of bases with quality values that are equal to or greater than 20 or 30, respectively.
2) Sequencing depth: Referring to the ratio of the total base numbers obtained from sequencing to the size of the target genome. If the gene size is 2M, and the sequencing depth is 10X, the total amount of data obtained amounts to 20M. Hence, the sequencing depth is calculated as Total Data (20M)/ Genome size (2M) = 10X.
3) Sequencing coverage: It defines the proportion of sequences obtained from sequencing in relation to the entire genome. Given the presence of complex structures, like sections with high GC content and repeat sequences in the genome, the sequences assembled after sequencing would not cover all areas. The regions devoid of sequences are termed "Gaps". For instance, if the sequencing coverage of a bacterial genome is 98%, 2% of the sequence region remains unsequenced.
Derivative Technologies for NGS
Although Next-Generation Sequencing (NGS) by itself only yields DNA sequences, it can generate an array of information, including targeted transcriptomes, methylomes, and metagenomes when combined with various sample processing techniques (such as in vitro transcription reversal, sequence capture, and bisulfite treatment). The correlation of these techniques is further facilitated by subsequent bioinformatics analyses. With the high-throughput precision offered by NGS, a vast range of clinical prospects is foreseeable. It presents potential applications in diverse clinical scenarios such as cancer screening, genetic testing, precision medicine and crop breeding, thus significantly broadening the extent of its usefulness (see Table 4).
Table 4. NGS sequencing derived technologies and their clinical applications
| Workflow | Sequencing Purpose | Application Scenarios |
| RNA-Seq | Reverse transcription of extracted transcriptome, followed by NGS sequencing of resulting cDNA, to obtain gene expression profiles within target cells. | Early screening for cancer, early diagnosis of metabolic diseases, companion diagnostics, etc. |
| ChIP-Seq | Utilizes chromatin immunoprecipitation (ChIP) to isolate DNA fragments bound to the target protein. After elution, NGS sequencing reveals the sequence of binding sites for the target protein. | Identification of binding sites for target transcription factors, discovery of biomarkers, and elucidation of gene regulatory networks. |
| Exon and Targeted Region Sequencing | Utilizes sequence capture technology to capture and enrich DNA from the entire genome's exons or other specific regions, followed by NGS. | Obtaining sequence information such as SNPs and structural variations in specific regions at a relatively low cost; used in neonatal genetic diagnosis and more. |
| Whole Genome Sequencing | Extracts genomic DNA from a mixed population for NGS sequencing, obtaining the core and variable genomes of the mixed species. | Analysis of sequencing sequences, including 16S rDNA, to gather information on the population and phenotype composition of the sample. Used in crop breeding, human microbiome transplantation, etc. |
| Methylation Sequencing | Applies sodium bisulfite treatment to DNA, converting unmethylated cytosine (C) to uracil (U). Whole-genome NGS sequencing is then performed. | Achieves single-base resolution at the whole-genome level, accurately assessing the methylation levels of individual C bases, and constructing detailed DNA methylation maps. Used in early cancer screening, early diagnosis of metabolic diseases, companion diagnostics, pharmacogenomics, etc. |
| Whole Genome Association Studies | Based on SNV analysis obtained from whole-genome sequencing, assesses its correlation with specific traits (diseases, immune responses) in specific populations. | Identifying SNVs that determine traits that are difficult to obtain through traditional molecular techniques. Used in estimating genetic disease risks, tumor detection, early cancer screening, precision medicine, etc. |
References:
- Maxam A M, Gilbert W. A new method for sequencing DNA. Proc Natl Acad Sci USA, 1977, 74(2): 560-564.
- Hillier L W, Marth G T. Quinlan A R, et al. Whole-ge nome sequencing and variant discovery in C. elegans. Nat Methodsl, 2008, 5: 183-188.
- D Y Zhang ,T X Zhang , G X Wang . Development and application of second- generation sequencing technology. Environmental Science & Technology, 2016, 39(9): 96-102.
- H CHEN , X F TAN. Excavation of Genie Resources Based on Next Generation Sequencing. Technologies Plant Physiology Journa,l 2014, 50(8): 1089-1095.
- Pacific Biosciences of California Inc. USA on world wide web URL [EB/OL]. 2017, 05, 20. http://www.pacb.oom/science/smrt-sequaicing/.
- Clarke J, Wu H C, Jayasinghe L, et al. Continuous base identification for single- molecule nanopore DNA sequencing. Nature Nanotechnology, 2009, 4(4): 265-270.
- P LU, J J JIN, Z F LI,P J CAO, et al. Genome assembly based on the Long-read sequencing technology and its application in tobacco. Tobacco Science& Technology, 2018, 02: 0087-0108.
- Haas B J, Zody M C. Advancing RNA-Seq analysis. Nat Biotechnol. 2010, 28(5): 421-423.
- Cock Peter J A, Fields Christopher J, Goto Naohisa The Sanger FASTQ file format for sequences with quality scores, and theSolexa/Illumina FASTQ variants. Nucleic Acids Research, 2009, 38(6): 1767-1771.
- L K Wang, Procession and application of RNA-Seq data [D]. 2012, 6.
- Z P Wu , X Wang , X G Zhang. Using non- uniform read distribution models to improve isoform expression inference in RNA-Seq. Bioinformatics (Online), 2010, 27(4): 502-508.
- Hansen Kasper D, Brenner Steven E, Dudoit Sandrine Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Research, 2010, 38(12), e131.
- He M, Y Wang , W P Hua , Y Z Zhang, Z Z Wang. De novo sequencing of Hypericum perforatum transcriptome to identify potential genes involved in the biosynthesis of active metabolites. PLoS ONE, 2012, 7(7): e42081.
- M Z Wang, B Wu, C H Chao and S F Lu.Identification of mRNA- like non-coding RNAs and validation of a mighty one named MAR in Panax ginseng. Journal of Integrative Plant Biology, 2015, 57(3): 256-270


