Next-Generation Sequencing of Fungal Genomes: Introductions, Technologies, and Applications
Inquiry >Mycology is a discipline focused on the research of fungi, which covers genetic and biochemical properties, taxonomy, and potential uses and hazards. Some fungi can be used as food resources, and some have promoted the development of the pharmaceutical industry (as a source for antibiotic drugs and statins). Meanwhile, there are also numerous fungi lead to infectious diseases, posing a threat to human, animals, and crops.
Despite that traditional morphology can be a straightforward and effective method with regard to initial inspections, it requires extensive mycological experience and might not always be accurate. The introduction of molecular methods, especially the next-generation sequencing (NGS) has made a major impact on mycology. These methods revolutionized mycological taxonomy and are still providing new information for determining major systematic divisions. More excitingly, these sequencing techniques help to detect mycobiota and uncover the molecular mechanisms of fungal phenotypes, which further facilitates the development and utilization of fungal resources as well as addresses fungi-caused problems.
NGS enhances the output of molecular data than previous technologies. The constantly improving capacity of the sequencing platform and the gradually decreasing cost make NGS a common technique for mycological labs. We summarize the NGS methodologies and their applications in mycology to give you a broader view of NGS in mycological studies.
Target-gene Amplicon Sequencing
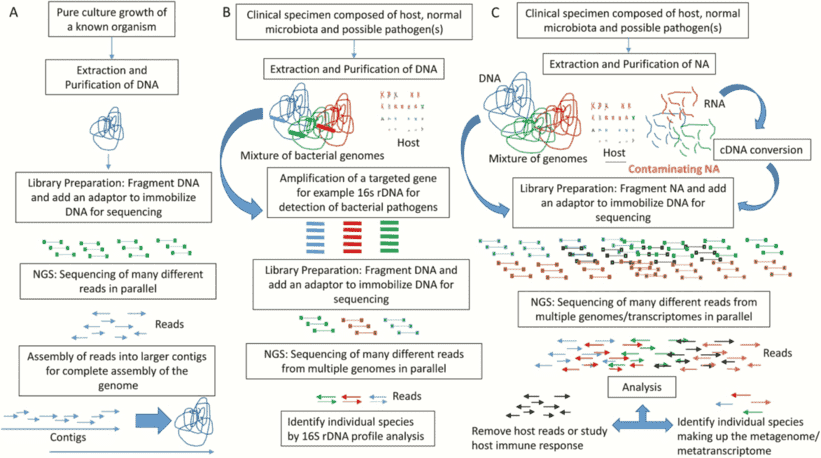
Amplicon sequencing is the most widely used high-throughput sequencing technique in microbial ecology. This culture-independent technology can characterize and identify mycobiome in complex and diverse environmental samples. There are three well studied regions that are commonly used in fugal metagenetic inspections, which are the ITS1/ITS2 region, 18S ribosomal small subunit RNA, and the D1/D2 domain of the 26S ribosomal large subunit (LSU) RNA. ITSs are regions that are ubiquitously found in bacteria, archaea, and fungi. ITSs generally show more variation than the ribosomal sequence, making them the most widely sequenced DNA region for identification and phylogenetic analysis of fungi, especially for the identification of yeasts and macrofungi, as well as for fungal diversity studies. Several fungi-specific primers are available for 18S rRNA gene sequences, with increasing public databases allowing for the discovery of new fungal groups and changes in fungal taxonomy. The primer pairs of fungus 18S rRNA gene, which should be carefully selected to meet the requirements of individual research projects, show great diversity in terms of species, fungal coverage, and amplification. The D1/D2 domain is a 500-600bp domain at the 5' end of LSU 26S rDNA, extensively used by many researchers in fungal sequencing.
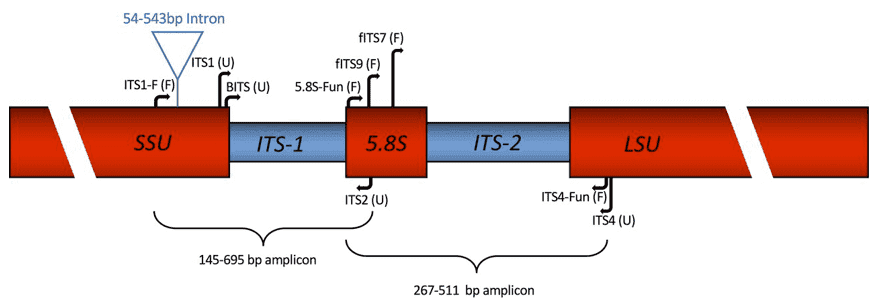 Figure 1. Fungal nuclear ribosomal ITS primer map. (Taylor 2016)
Figure 1. Fungal nuclear ribosomal ITS primer map. (Taylor 2016)
Methylation Sequencing with NGS
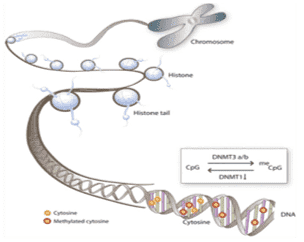
DNA methylation is a common epigenetic modification of DNA in eukaryotes that is crucial in cellular processes including genome regulation and development. Fungal DNA methylation, an intensively used method in fungal epigenomics profiling, is a stable marker of genomic defense because it is not present in structural genes and suppresses transposable factors and repetitive DNA sequences that undergo repeat-induced point mutations. NGS can analyze methyl markers at single-nucleotide and high-resolution. These methods vary in a number of aspects, such as DNA input, resolution, coverage, and bioinformatics analysis. Therefore, researchers need to have in-depth understanding of these techniques to be able to select the most feasible approach for their specific project.
Whole Genome Sequencing
Whole Genome Sequencing (WGS) has been widely used to provide detailed genetic information of fungi, including the relevance between phenotypic characters and genetic mechanisms. The rapid development of sequencing technologies and bioinformatic methods provide convenience for further investigation of the development, metabolism, phylogenetic taxonomy and evolution of fungi at the molecular level. There are several technical routes available for WGS, including Sanger sequencing and whole-genome shotgun sequencing. And two common assemble approaches for shotgun sequencing include de novo assembly and assembly by reference mapping. Mapping to a reference genome requires less contiguous reads and provides better identification of SNPs.
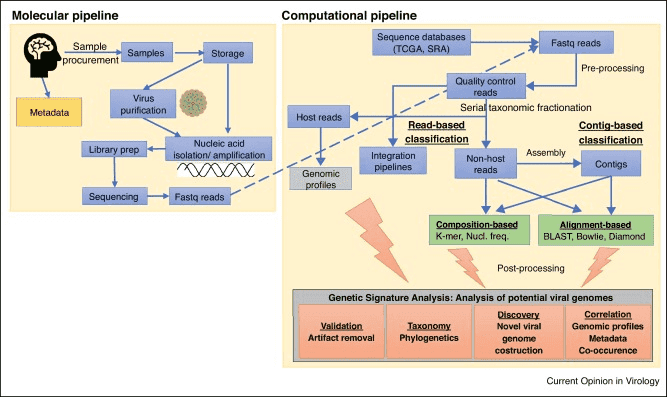
Shotgun Metagenomic sequencing
Shotgun metagenomic sequencing (or whole-genome shotgun sequencing, WGSS), can sequence billions of DNA base pairs in a single run without selective enrichment of a specific population. This method is not only capable of profiling microbial community composition and diversity but also provides the information to understand the functions encoded by the genomes of the microbiome. Shotgun metagenomic sequencing can provide a comprehensive view of microbial community within samples by being able to analyze DNA from different types of microbes including bacteria, archaea, and fungi.
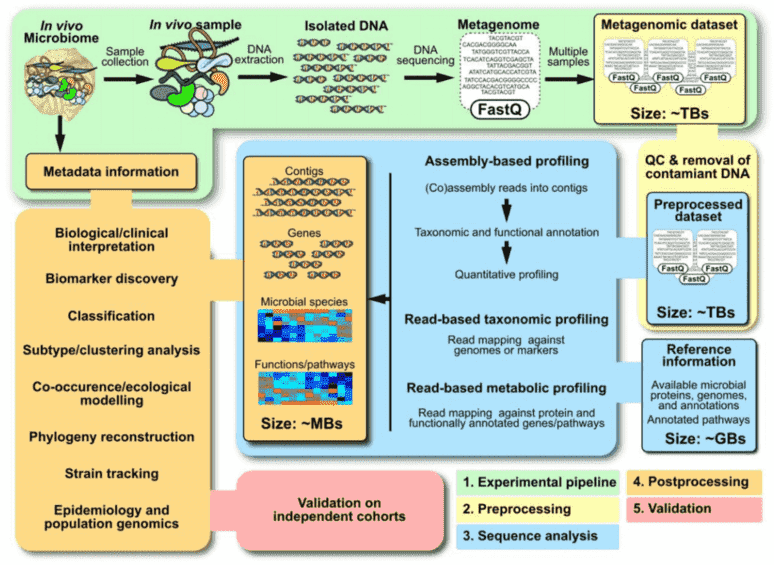 Figure 2. Summary of a metagenomics workflow. (Quince 2017)
Figure 2. Summary of a metagenomics workflow. (Quince 2017)
References
- McTaggart L; et al. Mycobiome sequencing and analysis applied to fungal community profiling of the lower respiratory tract during fungal pathogenesis. Front Microbiol. 2019, 10: 512. 2019.
- Forin N; et al. Next Generation Sequencing of Ancient Fungal Specimens: The Case of the Saccardo Mycological Herbarium. Frontiers in Ecology and Evolution. 2018, 6(129).
- Barros-Silva D; et al. Profiling DNA methylation based on next-generation sequencing approaches: new insights and clinical applications. Genes. 2018, 9(9):429.
- Quince C; et al. Shotgun metagenomics, from sampling to analysis. Nature biotechnology. 2017, 35(9):833.
- De Filippis F; et al. Different amplicon targets for sequencing-based studies of fungal diversity. Appl Environ Microbiol. 2017, 83(17).
- Zoll J; et al. Next-generation sequencing in the mycology lab. Current fungal infection reports. 2016, 10(2):37-42.
- Taylor DL; et al. Accurate Estimation of Fungal Diversity and Abundance through Improved Lineage-Specific Primers Optimized for Illumina Amplicon Sequencing. Applied and Environmental Microbiology. 2016, 82(24):7217-26.
- Lorenzen JM; et al. Epigenetic modifications in cardiovascular disease. Basic research in cardiology. 2012, 107(2):245.
- Zhang Y; Jeltsch A. The application of next generation sequencing in DNA methylation analysis. Genes. 2010, 1(1):85-101.