
 Sample Submission Guidelines
Sample Submission Guidelines
Metagenome Sequencing Reveal Changes in Intestinal Flora in COVID-19 Infections
What is COVID-19 infection's Gut Microbiota Metagenomics
Metagenomics technology bypasses the traditional method of microbial isolation and culture to extract total DNA directly from environmental samples, and obtains new functional genes and bioactive by constructing and screening metagenomic libraries, which include both culturable and unculturable microbial genetic information, thus increasing the chance of obtaining new bioactive.
Metagenomics is a new approach to microbial research in which the genomes of microbial populations in environmental samples are studied by functional gene screening and/or sequencing analysis, and can be used to analyze microbial diversity, population structure, evolutionary relationships, functional activity, and relationships with the environment.
The COVID-19 pandemic, caused by the novel coronavirus (Severe Acute Respiratory Syndrome Coronavirus 2 [SARS-CoV-2]), has elicited significant scientific research regarding its diverse clinical manifestations. Despite primarily affecting the respiratory system, COVID-19 has displayed an ability to infect various organs, notably the gastrointestinal (GI) tract alongside the pulmonary system. Reports suggest that gastrointestinal symptoms, often presenting as diarrhea, may precede respiratory symptoms in patients receiving treatment after viral clearance from the respiratory system, with the virus detected in fecal samples or rectal swabs for several days. Metagenomic sequencing can unveil alterations in the gut microbiota during COVID-19 infection, shedding light on the impact of COVID-19 infection on the composition and functionality of the gut microbiome, aiding in the understanding of the interactions between the virus and the host gut microbiota. Moreover, it can predict clinical outcomes, assisting in the identification of biomarkers for COVID-19 infection that may aid in early diagnosis, prognosis of disease progression, and development of personalized treatment plans.
Services you may interested in
Metagenomics Insights into COVID-19's Impact on Gut Microbiota
Shotgun Metagenomics reveals compositional changes in the gut microbiota of COVID-19 patients
In the article "Dissecting the role of the human microbiome in COVID-19 via metagenome-assembled genomes", published in Nature Communications in September, a total of 11,584 MAGs (metagenome-assembled genomes) and 5,403 non-redundant MAGs (nrMAGs) at the strain level were obtained from 514 nasal and fecal samples from COVID-19 patients, by metagenome assembly and binning techniques using published metagenome NGS data from six independent cohorts.
It was found that the strain abundance of many microbial species in the gut of COVID-19 patients was significantly reduced compared to normal subjects and that COVID-19 cases could be accurately distinguished from healthy controls based on gut microbiome characteristics and could predict the progression of COVID-19, along the lines shown below.
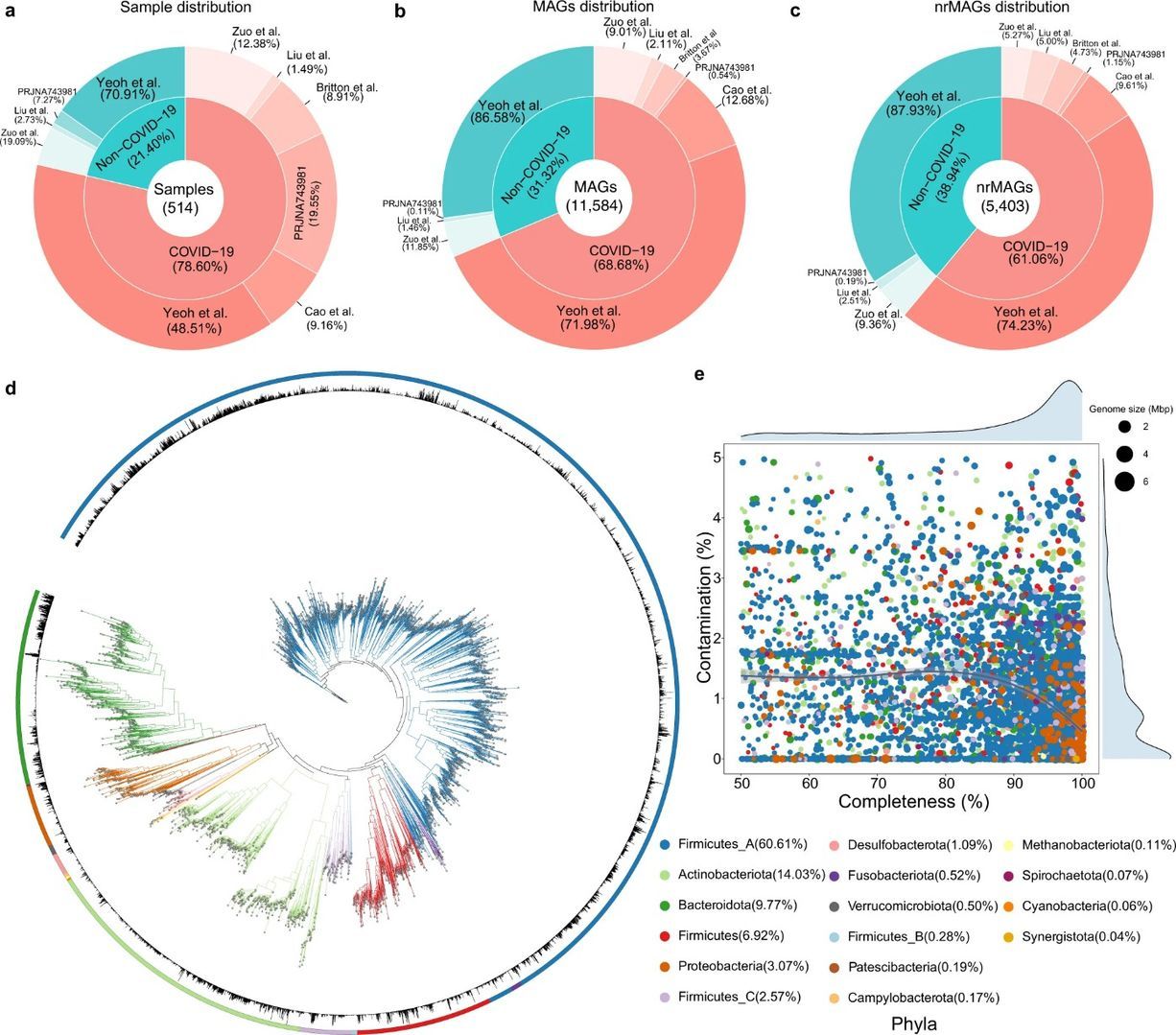 Figure 1. Reconstruction of MAGs from 514 COVID-19 related shotgun metagenomics sequencing data in the discovery cohorts (Ke S et al., 2022)
Figure 1. Reconstruction of MAGs from 514 COVID-19 related shotgun metagenomics sequencing data in the discovery cohorts (Ke S et al., 2022)
Analysis of hundreds of WMS sequencing data revealed that COVID-19 patients lost many strains (nrMAGs) for certain microbial species compared to non-COVID-19 controls. Through machine learning, the authors demonstrated the feasibility of accurately detecting COVID-19 from healthy controls based on gut microbiome signatures at nrMAG levels. Further understanding of the interactions between the human microbiome (including bacteria, fungi, and viruses) and SARS-CoV-2 infection, or other viral infections, may be discovered in the future with continued optimization of technology and data analysis methods.
In Maeda et al.'s (2022) study, gut mycobiota and microbiota were examined in severe COVID-19 patients, mild COVID-19 patients, and healthy individuals using metagenomic sequencing. The severe group exhibited lower mycobiota diversity with dominance of Candida albicans, while microbial diversity was reduced with increased Enterococcus and Lactobacillus, and decreased Faecalibacterium and Bacteroides abundance. No significant differences were found between mild COVID-19 patients and healthy individuals. Candida abundance correlated positively with Enterococcus. After severe patients recovered, microbiota alterations persisted, but mycobiota diversity rebounded to levels akin to mild patients and healthy individuals.
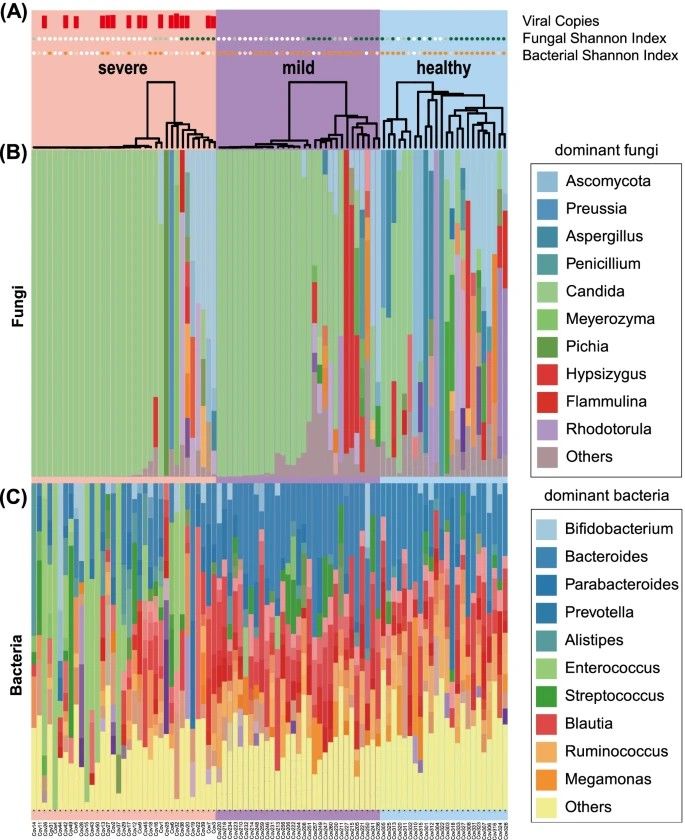 Figure 2. Altered composition of the gut mycobiota and microbiota in patients with COVID-19. (Maeda et al., 2022)
Figure 2. Altered composition of the gut mycobiota and microbiota in patients with COVID-19. (Maeda et al., 2022)
Cao et al. (2021) conducted a study in Beijing utilizing metagenomic analysis to explore the gut virome and bacteriome in 13 COVID-19 patients compared to 5 healthy controls. Their investigation revealed bacterial dysbiosis in COVID-19 patients, characterized by diminished microbial diversity and viral shifts. Severe cases of COVID-19 displayed an elevated presence of opportunistic pathogens and a decline in butyrate-producing bacteria. These observations were corroborated in a mouse model of COVID-19, which exhibited variations in the expression of immune and infection-related genes within gut epithelial cells. The findings propose a significant impact of SARS-CoV-2 infection on the microbiome, with potential implications for disease severity and recovery.
Zuo et al. (2020) employed shotgun metagenomic sequencing to scrutinize the fecal microbiomes of 15 COVID-19 patients in Hong Kong during hospitalization, juxtaposed with 6 individuals with community-acquired pneumonia and 15 healthy controls. Their investigation unveiled noteworthy and enduring modifications in the gut microbiome of COVID-19 patients, manifested by an upsurge in opportunistic pathogens and a decline in beneficial commensals, which exhibited a correlation with disease severity and fecal SARS-CoV-2 load. These observations propose that alterations to the gut microbiota could potentially ameliorate the severity of COVID-19.
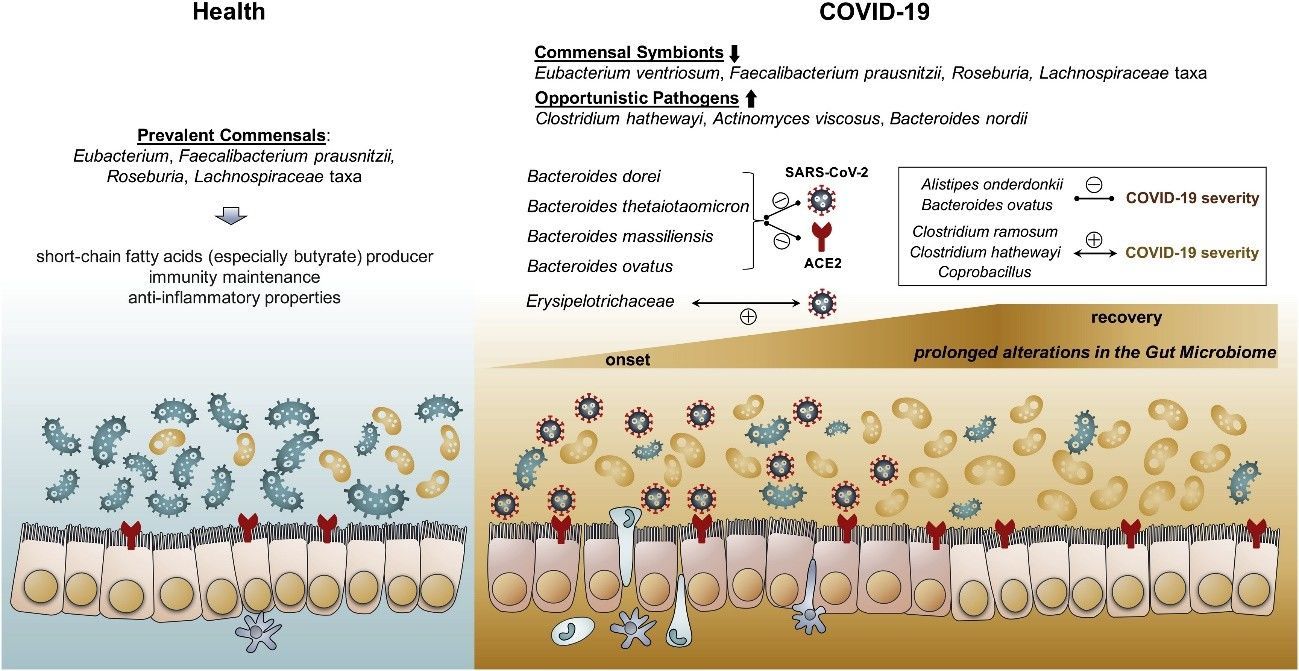 Figure 3. Schematic summary of the gut microbiome alterations in COVID-19. (Zuo et al., 2020)
Figure 3. Schematic summary of the gut microbiome alterations in COVID-19. (Zuo et al., 2020)
Li et al. (2021) employed shotgun metagenomic sequencing to investigate the gut microbiota of 47 COVID-19 patients in comparison to 19 healthy controls. They detected four distinct microorganisms exclusive to COVID-19 patients and noted considerable variations in the abundance of diverse bacterial species. COVID-19 patients exhibited elevated levels of Bacteroides stercoris, B. vulgatus, B. massiliensis, among others, while species like Clostridium nexile and Streptococcus salivarius were diminished. Moreover, a significant reduction in the butyrate-producing Roseburia inulinivorans and an increase in Paraprevotella sp. and Streptococcus thermophilus were observed. The research identified 30 KEGG orthology modules displaying significant alterations and 15 microbial markers with robust diagnostic potential for discriminating COVID-19 cases. Associations between specific bacterial species and clinical parameters were established, along with a shift in the Bacteroidetes to Firmicutes ratio. These findings suggest that shifts in gut microbiota composition might influence the severity of COVID-19, and microbial markers hold promise for improving COVID-19 diagnostics.
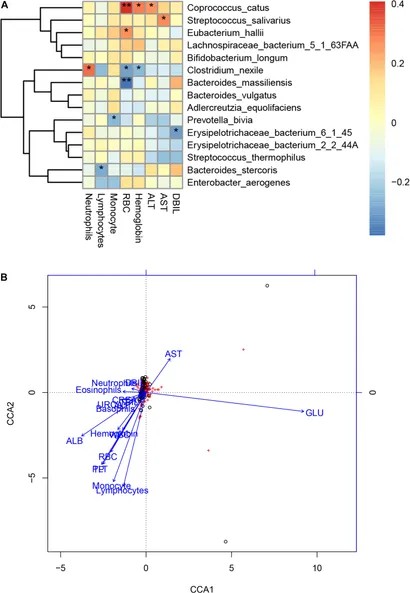 Figure 4. Associations between the gut microbiome and clinical indexes of COVID-19. (Li et al., 2021)
Figure 4. Associations between the gut microbiome and clinical indexes of COVID-19. (Li et al., 2021)
How to Analyze Metagenomic Data
Using the data analysis in the article "Dissecting the role of the human microbiome in COVID-19 via metagenome-assembled genomes" as an example to understand metagenomic data analysis:
1. metagenome assembly and binning
The authors used metaWRAP, an analysis process that integrates quality control, splicing, boxing, purification, evaluation, species annotation, abundance estimation, functional annotation and visualization, to process the raw sequencing data, incorporating over 140 tools. The detailed process is as follows. (a). QC and removal of host contamination of raw data; (b). Assembly of the decontaminated data using the metaSPAdes tool in the metaWRAP; (c). binning using MaxBin2, metaBAT2 and CONCOCT software, and purifying the binning results using the bin_refinement module, and finally assessing the contamination rate and completeness of the results using CheckM.
2. Clustering and dereplication of MAGs
The MAGs were clustered on species-level genome bins (SGBs) using the software dRep, and then the MAGs were taxonomically annotated based on the Genome Taxonomy database using the software GTDB-Tk.
3. Calculation of species abundance and phylogenetic tree
The abundance of each nrMAGs was calculated using the software Salmon, and evolutionary trees were constructed using PhyloPhlAn and embellished using iTOL.
4. Genome annotation of nrMAGs
The genomes obtained from bining were genetically predicted using Prokka software, and the genes were functionally annotated using MicrobeAnnotator and the completeness of each database annotation was assessed. Finally, functional analysis was performed using HUMANN3.
5. Statistical analysis
Alpha and Beta microbial diversity were calculated using the R's vegan package, and random forest regression analysis was performed using R' random Forest package.
Metagenomic data analysis entails a series of pivotal steps aimed at unraveling the taxonomic and functional landscapes of intricate microbial communities. Initially, raw shotgun metagenomic data is acquired from a diverse array of microbial populations and subsequently subjected to rigorous quality control processing to ensure data precision. Following this, the data undergoes taxonomic profiling through marker gene analysis, elucidating the species composition and phylogenetic diversity within the community. Binning analysis plays a crucial role in categorizing these sequences into bins that represent individual genomes or operational taxonomic units.
Assembly analysis is essential for reconstructing fragmented genomes or sequencing reads into contigs, facilitating the identification of novel taxa and genes. Gene prediction and functional annotation decode genomic information, revealing biological roles and pathways prevalent in the community. Additionally, comparative analysis of metagenomes enables evaluation of community similarities, assessment of environmental impacts on community structure, and identification of key taxa or functions distinguishing microbial communities. This comprehensive analytical approach provides insights into microbial ecosystem characteristics, potential biomarkers, and functional roles within diverse metagenomic datasets, advancing our understanding of complex microbial communities.
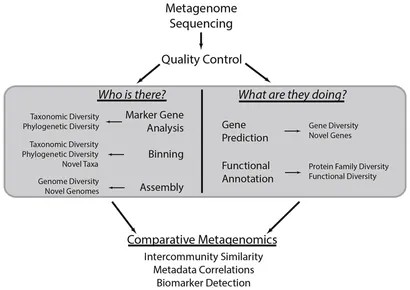 Figure 5. Common metagenomic analytical strategies. (Sharpton, 2014)
Figure 5. Common metagenomic analytical strategies. (Sharpton, 2014)
Summary
In the exploration of the gut microbiota of COVID-19 patients, a multitude of researchers have turned to metagenomics. Their endeavors have uncovered notable shifts within the gut microbiome of individuals afflicted with COVID-19, characterized by a reduction in microbial diversity and notable alterations in specific bacterial populations. These observed variances serve as critical discriminators between COVID-19 cases and their healthy counterparts, offering predictive insights into disease progression. Furthermore, the incorporation of metatranscriptomics into these investigations provides a deeper comprehension of the gut microbiota in COVID-19 patients, shedding light on the influence of these microbial entities on the disease via analyses of functional pathways and gene expression patterns.
Despite these advancements, our current scientific understanding of the intricate interplay between gut microbiota and COVID-19 remains incomplete and dynamically evolving. Hence, the judicious selection and proficient application of computational tools and methodologies become imperative in effectively interrogating gut microbiota. This strategic approach represents a pivotal step towards unraveling the most pertinent diagnostic microbial profiles and crafting precise antiviral or prophylactic microbiome interventions. Such endeavors not only enrich our comprehension of disease mechanisms but also foster the emergence of novel perspectives and methodologies in the diagnosis and treatment of COVID-19.
Recommended Services
CD Genomics provides Viral Metagenomic Sequencing and Metagenomic Shotgun Sequencing based on Illumina, Nanopore sequencing and PacBio SMRT sequencing platforms, helping you analyze various gut microbial samples, study the relationship between the microbiome and the host, and analyze the function of microorganisms at the genetic level.
You may also be interested in the following article:
Next Generation Sequencing for COVID-19
Bioinformatics Analysis of Viral Metagenomic Sequencing
Applications of Metagenomics in Biotechnology and Health Care
16S rRNA Sequencing vs. Metagenome Sequencing: A Guide for Selection
References:
- Ke S, Weiss S T, Liu Y Y. Dissecting the role of the human microbiome in COVID-19 via metagenome-assembled genomes. Nature communications, 2022, 13(1): 1-15.
- Maeda Y, Motooka D, Kawasaki T, et al. Longitudinal alterations of the gut mycobiota and microbiota on COVID-19 severity. BMC infectious diseases, 2022, 22(1): 572.
- Cao J, Wang C, Zhang Y, et al. Integrated gut virome and bacteriome dynamics in COVID-19 patients. Gut microbes. 2021, 13(1):1887722.
- Li S, Yang S, Zhou Y, et al. Microbiome profiling using shotgun metagenomic sequencing identified unique microorganisms in COVID-19 patients with altered gut microbiota. Frontiers in Microbiology. 2021, 12:712081.
- Zuo T, Zhang F, Lui GC, et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology. 2020, 159(3):944-55.
- Sehli S, Allali I, Chahboune R, et al. Metagenomics approaches to investigate the gut microbiome of COVID-19 patients. Bioinformatics and Biology Insights. 2021, 15:1177932221999428.
- Sharpton, T.J. An introduction to the analysis of shotgun metagenomic data. Frontiers in plant science, 2014, 5, p.86894.


