
 Sample Submission Guidelines
Sample Submission GuidelinesWe use cookies to understand how you use our site and to improve the overall user experience. This includes personalizing content and advertising. Read our Privacy Policy
Accept Cookies

November 1, 2019
Xi Wang, Xintian You, Julian D. Langer, Jingyi Hou, Fiona Rupprecht, Irena Vlatkovic, Claudia Quedenau, Georgi Tushev, Irina Epstein, Bernhard Schaefke, Wei Sun, Liang Fang, Guipeng Li, Yuhui Hu, Erin M. Schuman & Wei Chen
Nature Communications volume 10, Article number: 5009 (2019)
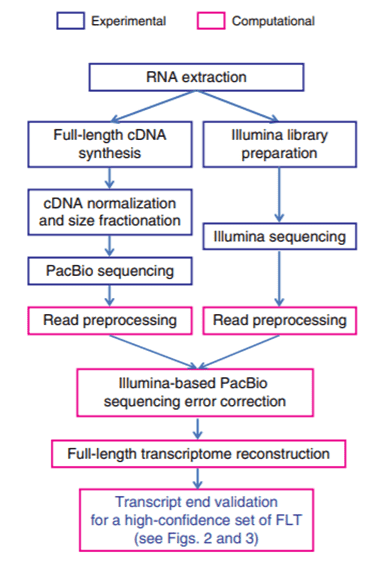
The flow chart of the hybrid sequencing workflow, including the experimental procedures (blue rectangles) comprised of PacBio sequencing and Illumina sequencing, and the computational components (pink rectangles) for Illumina-based PacBio error correction, followed by full-length transcriptome reconstruction, transcript end validation, and isoform diversity characterization
Abstract
Gene annotation is a critical resource in genomics research. Many computational approaches have been developed to assemble transcriptomes based on high-throughput short-read sequencing, however, only with limited accuracy. Here, we combine next-generation and third-generation sequencing to reconstruct a full-length transcriptome in the rat hippocampus, which is further validated using independent 5´ and 3´-end profiling approaches. In total, we detect 28,268 full-length transcripts (FLTs), covering 6,380 RefSeq genes and 849 unannotated loci. Based on these FLTs, we discover co-occurring alternative RNA processing events. Integrating with polysome profiling and ribosome footprinting data, we predict isoform-specific translational status and reconstruct an open reading frame (ORF)-eome. Notably, a high proportion of the predicted ORFs are validated by mass spectrometry-based proteomics. Moreover, we identify isoforms with subcellular localization pattern in neurons. Collectively, our data advance our knowledge of RNA and protein isoform diversity in the rat brain and provide a rich resource for functional studies.
More info at: https://www.nature.com/articles/s41467-019-13037-0

