
 Sample Submission Guidelines
Sample Submission Guidelines
MeDIP‑Seq / hMeDIP‑Seq (DNA Methylation & Hydroxymethylation Profiling)
CD Genomics offers MeDIP‑seq for genome‑wide methylation profiling and hMeDIP‑seq to selectively capture 5‑hydroxymethylcytosine (5hmC), enabling parallel analysis of multiple cytosine modifications.
Introduction to MeDIP seq and hMeDIP seq
DNA methylation refers to the postreplicative maintenance or de novo addition of a methyl group to the carbon-5 position of the cytosine pyrimidine ring by DNA methyltransferases. In mammals, DNA methylation typically occurs in CG (mCG) and non-CG (mCHG or mCHH, collectively referred to as mC) contexts. mCG alone accounts for approximately 60–80% of all CG dinucleotides. In addition to 5-methylcytosine (5mC), several cytosine modifications have been identified, including 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, and 5-carboxylcytosine. Among them, 5hmC is structurally similar to 5mC but much less abundant, and is generated through the oxidation of 5mC by the TET family of dioxygenases.
DNA methylation and hydroxymethylation are both crucial epigenetic marks implicated in gene regulation, embryonic development, cellular differentiation, and genome stability. Genome-wide analysis of both 5mC and 5hmC provides critical insight into epigenetic dynamics, particularly in developmental and stress response studies.
MeDIP‑seq couples immunoprecipitation with high-throughput sequencing to enrich and profile 5mC across the genome. It utilizes anti‑5mC antibodies to selectively capture DNA fragments containing methylated cytosines (in mCG and mCH contexts). These enriched fragments are sequenced, and their read distribution can be used to estimate relative methylation levels.
To distinguish and specifically profile 5hmC, CD Genomics also offers hMeDIP‑seq, which employs 5hmC‑specific antibodies to enrich hydroxymethylated DNA fragments. This complementary approach allows researchers to separately profile 5mC and 5hmC modifications in parallel, enhancing the resolution of epigenetic mapping across diverse sample types. Both MeDIP‑seq and hMeDIP‑seq are non-destructive, conversion-free methods that do not rely on bisulfite treatment or uracil-tolerant polymerases, and are compatible with low-input samples.
Advantages of MeDIP-Seq / hMeDIP-Seq
- Can target mC, mCG, or hmC
- Whole genome or any regions of interest
- Near-unbiased and hypothesis
- Epigenetic biomarker discovery
- Single-nucleotide resolution and cost-efficiency
- Low input DNA requirement
Applications of MeDIP-Seq / hMeDIP-Seq
- Epigenetic Heterogeneity
- Environment and Epigenetics
- Genetic Expression Regulation
- Disease Research
- Genetic Imprinting
- Embryonic Development
- Detection of DNA methylation-enriched regions in disease samples and inference of candidate gene sets whose expression is suppressed in the samples.
- Monitoring the dynamic DNA methylation patterns at different stages of disease occurrence and development, particularly in lesion sites, to screen for epigenetic markers that help define the extent of disease progression.
- Comparative analysis of the differences in signal and location distribution of DNA methylation regions between disease and normal samples, identification of disease-specific DNA methylation regions, and observation of the genes surrounding these regions to narrow down the list of candidate genes related to the disease.
- Mapping of hydroxymethylation (5hmC) landscapes in tissues, cell differentiation, or stress-response models using hMeDIP-seq.
MeDIP-Seq / hMeDIP-Seq Workflow
The general workflow for MeDIP sequencing is outlined below. Briefly, the extracted DNA is fragmented, denatured, ligated with adaptor and captured using the antibody directed against 5-methylcytosine (for MeDIP-seq) or 5-hydroxymethylcytosine (for hMeDIP-seq), followed by library preparation and sequencing on Illumina platforms.

Service Specification
Sample Requirements
|
|
 Click |
Sequencing Strategies
|
 |
Data Analysis We provide multiple customized bioinformatics analyses:
|
Analysis Pipeline
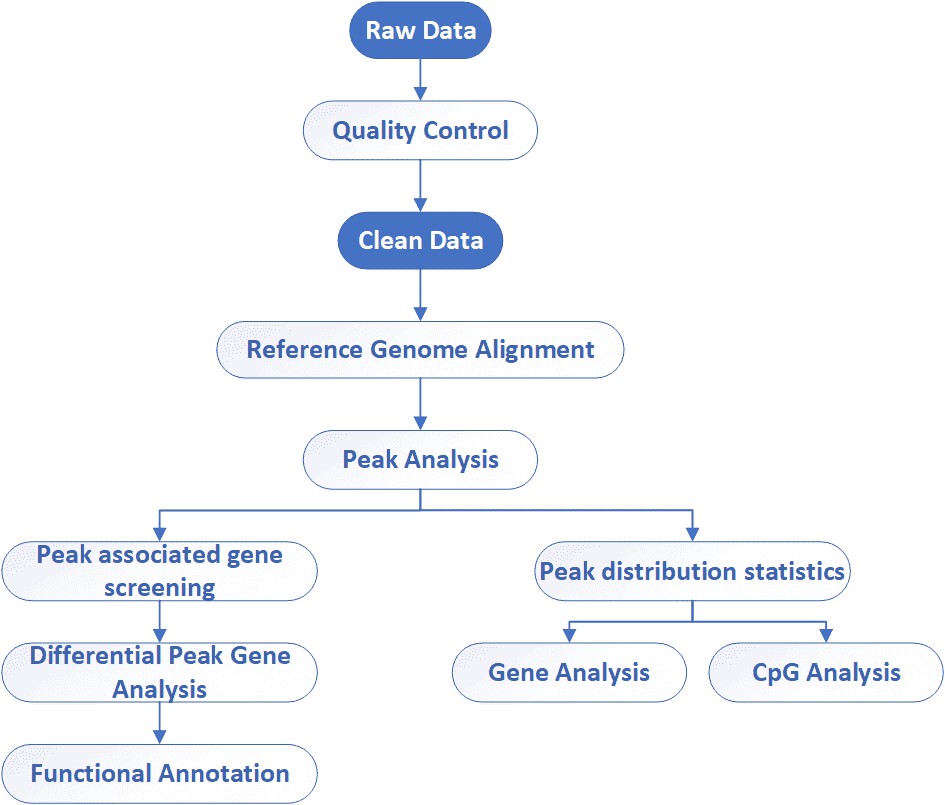
Deliverables
- The original sequencing data
- Experimental results
- Data analysis report
- Details in MeDIP-Seq / hMeDIP-Seq for your writing (customization)
With professional bioinformatics capability, CD Genomics offers high-quality MeDIP-Seq as an end-to-end, genome-wide epigenetic service to identify differentially methylated regions, and ultimately help to expedite epigenetic research. If you have additional requirements or questions, please feel free to contact us.
Reference:
- Taiwo O, Wilson G A, Morris T, et al. Methylome analysis using MeDIP-seq with low DNA concentrations. Nature protocols, 2012, 7(4): 617.
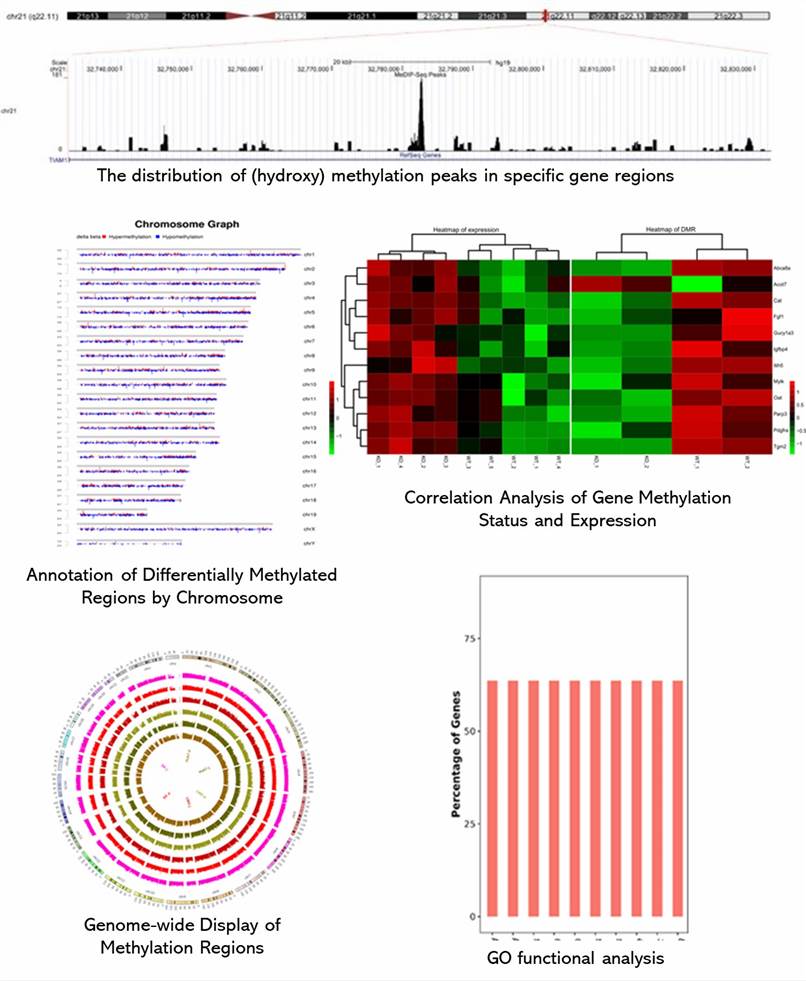
Demo Results
Partial results are shown below:
MeDIP‑Seq / hMeDIP‑Seq FAQs
1. Compared with other methods, what are the advantages of MeDIP sequencing?
Currently, sequencing-based approach for methylome analysis can be classified into bisulfite conversion-based and enrichment-based approaches. Bisulfite conversion-based methods include whole-genome or targeted bisulfite sequencing or reduced representation bisulfite sequencing (RRBS). Although bisulfite conversion-based methods are considered to be the gold standard of DNA methylation analysis at single-base resolution, they cannot distinguish between mC and hmC. Additionally, whole genome bisulfite sequencing is expensive for application on large sample sizes and by smaller research groups. And RRBS can only provide limited genome coverage (5-10%) and is centered on CpG island and promoter regions.
Besides MeDIP sequencing, enrichment-based technologies also include MBD-seq, which uses the methyl-binding protiens MBD2 and MDB3L1, and methylCap-seq that use methyl-binding domain of MECP2 for methyl capture. But MBD-seq and methylCap-seq are restricted to the analysis of mCG and whole-genome protocols often require high concentrations of genomic DNA (more than 1,000 ng). Therefore, MeDIP is a versatile, accurate, and costly method with a low input DNA requirement and is applicable to a wide range of samples and studies.
2. What are the requirements for MeDIP sequencing samples?
MeDIP sequencing samples require reference genome or sequences for alignment, and the assembled results directly affect the accuracy of data analysis. Reference genome from closely related species or assembled results from transcriptome can also be used despite the loss of partial methylation information.
3. What factors can affect the results of MeDIP-seq?
Every process involved in MeDIP-seq may affect the results, especially the immunoprecipitation and PCR. Immunoprecipitation is the process to enrich methylated regions, and reaction conditions may influence the enrichment results. If insufficient DNA is recycled, PCR is used to scale it up, which can biase results. Furthermore, contamination should be avoided throughout the whole course.
MeDIP‑Seq / hMeDIP‑Seq Case Studies
Epigenetic alterations in TRAMP mice: epigenome DNA methylation profiling using MeDIP-seq
Journal: Cells & Bioscience
Published: 12 January 2018
Abstract
The authors profiled the methylome of the mouse prostate (TRAMP) cancer model and to analyze the crosstalk among targeted genes and the related functional pathway. By utilizing MeDIP sequencing, they analyzed DNA methylation profiles and performed relevant informatics analyses, which could provide strategies for prevention and treatment approaches for prostate cancer.
Materials & Methods
- TRAMP mice and C57BL/6 mice
- Genomic DNA extraction
- MeDIP-seq
- Illumina HiSeq2000
- RT-PCR
- Methylation-specific PCR
- Alignment with the reference mouse genome
- Canonical pathways
- Diseases and function and network analysis
Results
1. MeDIP-seq results comparison
In total, 2147 genes between TRAMP and control animals showed a significant change in methylated peaks. Compared with the control, significantly increased methylation of 1042 genes and significantly decreased methylation of 1105 genes were observed in TRAMP. Four genes of interest, DYNC1I1, SLC1A4, XRCC6BP1, and TTR were analyzed by IGV. TRAMP mice showed increased methylation ratio of DYNC111 and SLC1A4, and decreased methylation ratio of TTR and XRCC6BP1, which was in accordance with the MeDIP-seq results.
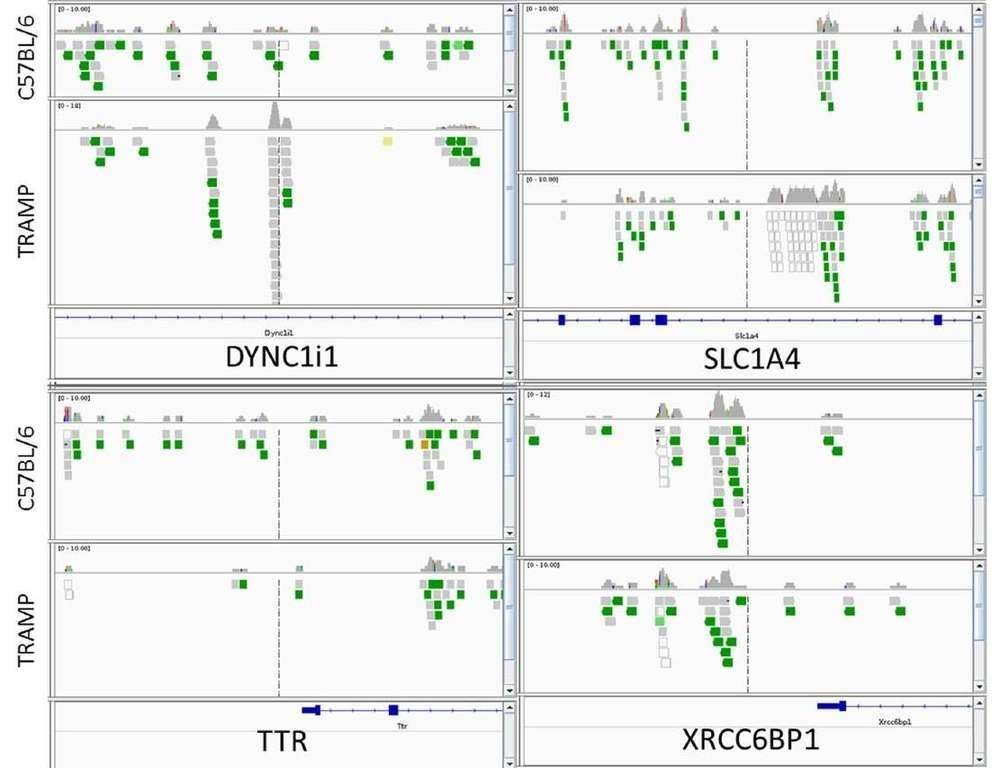 Figure 1. integrative genomics viewer visualization of the aligned reads' distribution against reference genome for four target genes, DYNC1I1, SLC1A4, XRCC6BP1, and TTR.
Figure 1. integrative genomics viewer visualization of the aligned reads' distribution against reference genome for four target genes, DYNC1I1, SLC1A4, XRCC6BP1, and TTR.
2. qPCR validation of selected gene expression
The expression levels of CRYZ, DYNC1I1, HNMT, SLC1A4, and TTR were measured in both TRAMP and wide type group (Figure 2). Among them, TTR expression was increased by 9.05-fold over WT. And the expression levels of TTR were significantly higher in prostate cancer tissue than in normal and benign prostate hyperplasia tissue. Furthermore, they found decreased methylation in promoter region of TTR but increased gene expression. In contrast, DNA methylation in the gene body or downstream may or may not follow a reciprocal relationship.
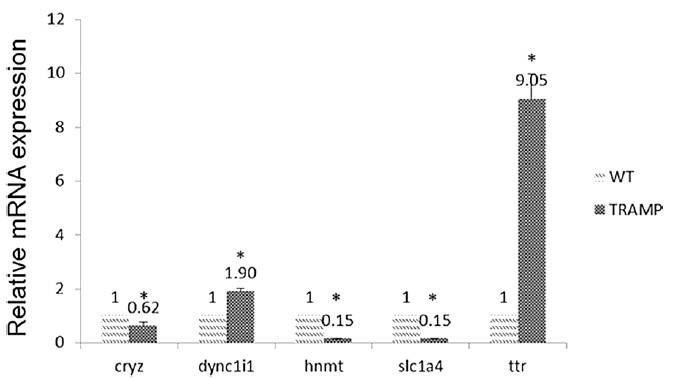 Figure 2. Comparison of miRNA expression of CRYZ, DYNC1I1, HNMT, SLC1A4, and TTR among wide type and TRAMP mice prostate samples.
Figure 2. Comparison of miRNA expression of CRYZ, DYNC1I1, HNMT, SLC1A4, and TTR among wide type and TRAMP mice prostate samples.
3. Canonical pathway, diseases and functions and network analyses.
The 2147 genes with significant change in methylation were analyzed. The cancer-related networks accounted for the majority (Table 2), which suggested that the difference between the TRAMP and control lay in organ development and cancer development. The most associated disease, cancer, gastrointestinal disease, organismal abnormalities, reproductive system disease and dermatological diseases were ranked within the top five (Figure 3).
Table 1. Top ten altered canonical pathways.
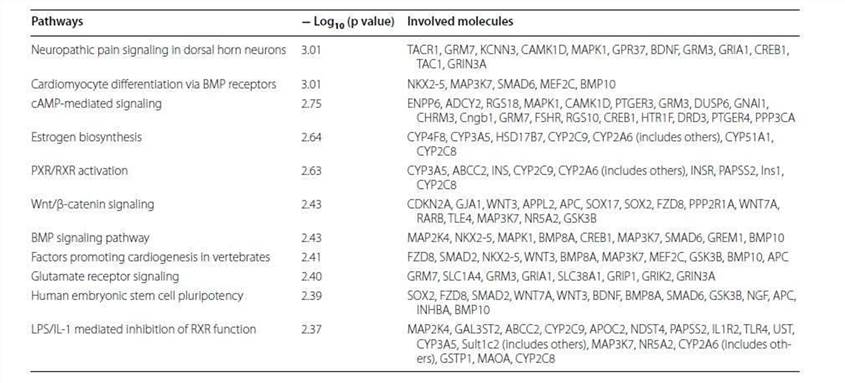
Table 2. Top networks analyzed.
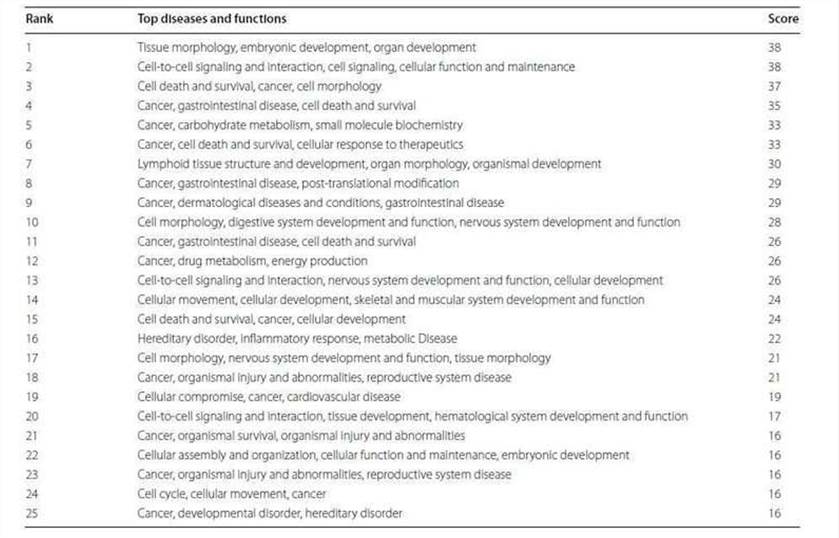
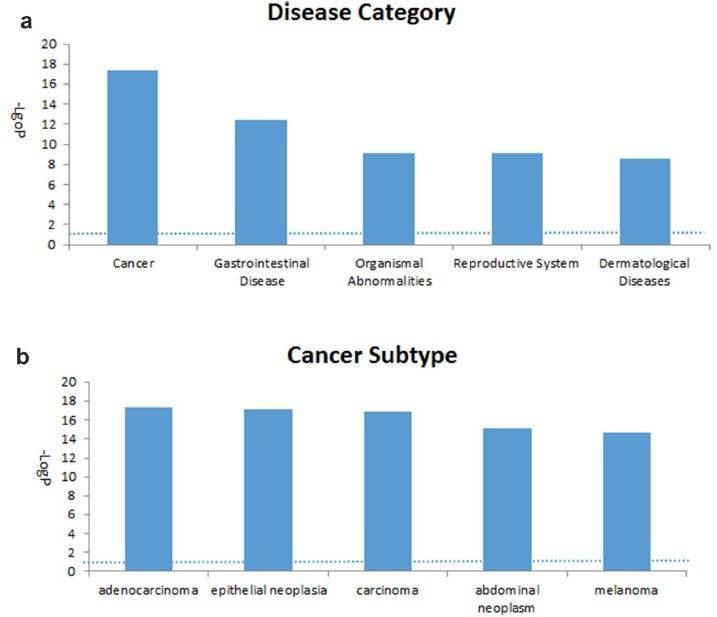 Figure 3. Top five associated disease categories (a) and top five cancer subtypes (b) analyzed.
Figure 3. Top five associated disease categories (a) and top five cancer subtypes (b) analyzed.
Discussion
Analysis of canonical pathway would provide information for the development of new therapeutic targets. As shown in Figure 4, the genes with significant methylation in the top canonical pathway was the neuropathic pain signaling pathway, which is consistent with the former finding that the most common malignancy in TRAMP is of neuroendocrine origin. Table 3 lists the genes involved in this pathway that showed modified methylation. CREB was found to be closely related to cellular proliferation, differentiation and adaptive responses in the neuronal system, and methylation of the CREB1 gene was found to be decreased by 2.274 in TRAMP. Furthermore, CREB was also found to regulate other carcinogenesis pathways. All of these data indicate that CREB is highly linked with cancer therapy and may be new strategy for prostate cancer prevention and therapy.
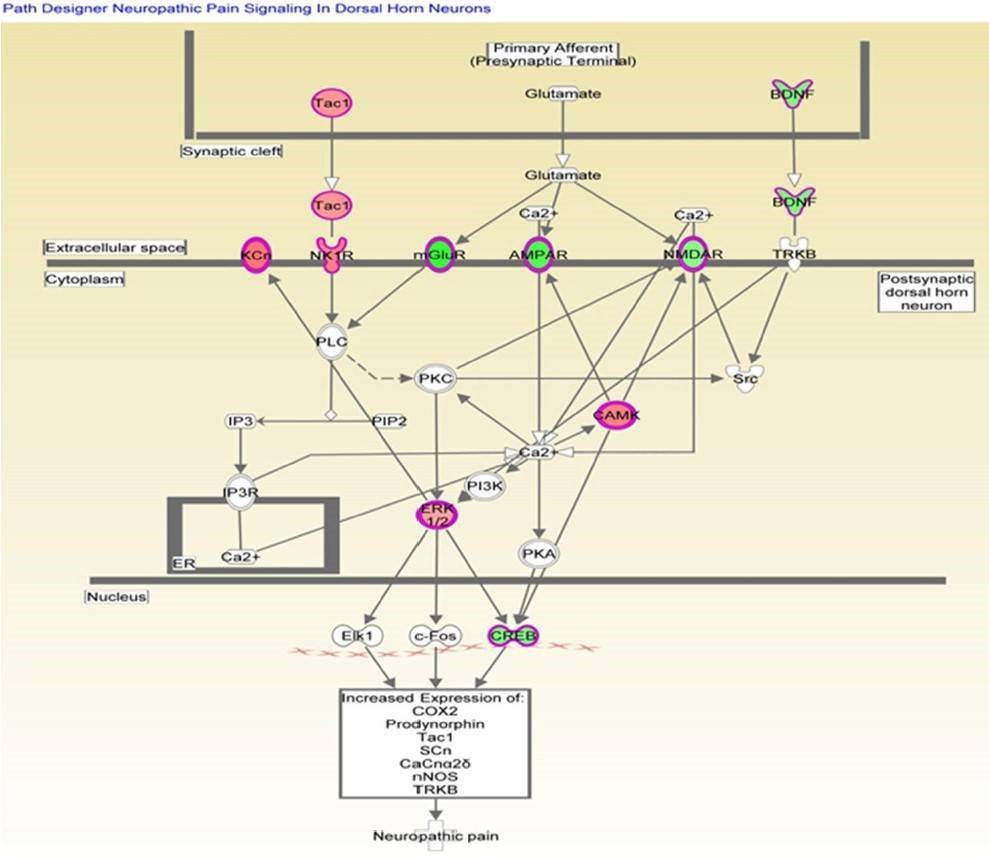 Figure 4. Genes mapped to the canonical neuropathic pain signaling pathway.
Figure 4. Genes mapped to the canonical neuropathic pain signaling pathway.
Table 3. Altered methylation genes mapped to the neuropathic pain signaling pathway.
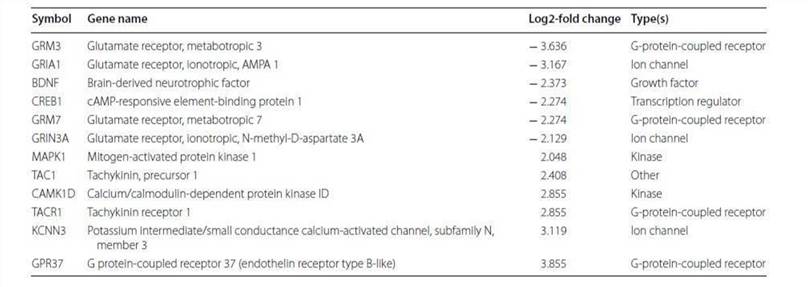
Reference:
- Li W, Huang Y, Sargsyan D, et al. Epigenetic alterations in TRAMP mice: epigenome DNA methylation profiling using MeDIP-seq. Cell & bioscience, 2018, 8(1): 3.
Related Publications
Here are some publications that have been successfully published using our services or other related services:
Restriction endonuclease cleavage of phage DNA enables resuscitation from Cas13-induced bacterial dormancy
Journal: Nature microbiology
Year: 2023
IL-4 drives exhaustion of CD8+ CART cells
Journal: Nature Communications
Year: 2024
High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
Journal: International Journal of Molecular Sciences
Year: 2024
KMT2A associates with PHF5A-PHF14-HMG20A-RAI1 subcomplex in pancreatic cancer stem cells and epigenetically regulates their characteristics
Journal: Nature communications
Year: 2023
Cancer-associated DNA hypermethylation of Polycomb targets requires DNMT3A dual recognition of histone H2AK119 ubiquitination and the nucleosome acidic patch
Journal: Science Advances
Year: 2024
Genomic imprinting-like monoallelic paternal expression determines sex of channel catfish
Journal: Science Advances
Year: 2022
See more articles published by our clients.