
 Sample Submission Guidelines
Sample Submission Guidelines
Epigenomics Sequencing (DNA/RNA Methylation Analysis)
CD Genomics is offering platforms for genome-wide epigenomics analysis, each designed to accommodate a wide range of sample types and suit your specific research needs, allowing researchers to look at epigenetic alterations easily. This information will help us not only understand the role of DNA methylation but also identify targets for therapeutic treatment.
What is Epigenetic Modifications
Epigenetic modifications are reversible modifications that affect gene expression without altering the DNA sequence and can be inherited during cell division. Two of the most characterized epigenetic modifications are DNA methylation and chromatin modification. Epigenetic modifications play important roles in gene expression and regulation, and are involved in numerous cellular processes such as in differentiation, development and tumorigenesis. DNA methylation is most frequently observed at the C5 position of cytosine followed by guanine (CpG site) in vertebrates, or non-CpG sites such as CHG and CHH in plants or mammalian embryonic stem cells. DNA methylation is established and maintained by DNA methyltransferases (DNMT1, DNMT3a, and DNMT3b).
Epigenomics can be divided into two main categories:
1. Epigenome: The epigenome encompasses a vast array of chemical modifications occurring on DNA and histones within the chromatin, in addition to structural changes in chromatin itself. These modifications, independent of the underlying genomic sequence, provide critical regulatory information. The following are key components of the epigenome:
(1) DNA Modifications: Includes 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC).
(2) Histone Modifications: Encompasses modifications such as H3K27me3, H3K4me3, and H3K27ac.
(3) Chromatin Level Changes: Involves alterations mediated by DNA-binding proteins such as transcription factors.
2. Epitranscriptome: This term broadly refers to all post-transcriptional modifications that do not alter the RNA sequence itself. Currently, over 100 different chemical modifications have been identified on RNA, including N6-methyladenosine (m6A), N1-methyladenosine (m1A), and pseudouridine (Ψ).
DNA Methylation Sequencing Technologies
DNA methylation information may be lost during standard molecular biology manipulations, such as molecular cloning in bacteria and PCR, due to lack of maintenance of DNA methyltransferases. Several techniques include methylated DNA immunoprecipitation (MeDIP), bisulfite sequencing (BS-seq), and reduced representation bisulfite sequencing (RRBS) have been proposed to preserve DNA methylation information and simultaneously transform it into quantitative and measurable signals. By combining with high-throughput sequencing, these techniques have provided comprehensive and reliable genome-wide information regarding DNA methylation. A brief outline and workflow of different NGS-based DNA methylation sequencing technologies is shown in below Figure 1.
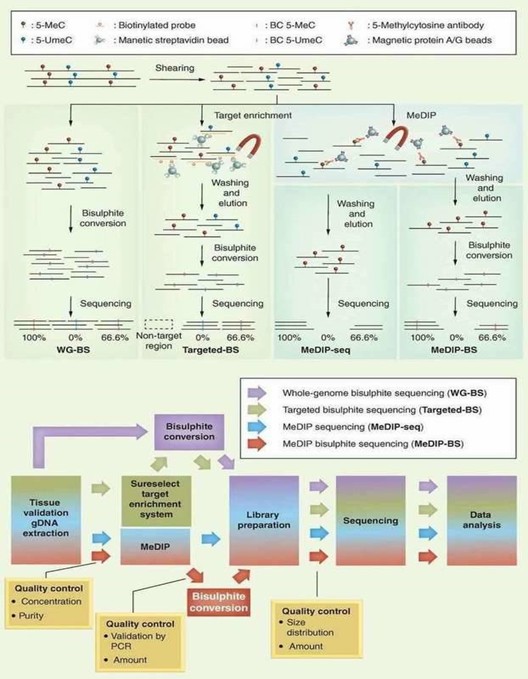 Figure 1. Different NGS-based DNA methylation analysis methods (Jeong et al., 2016).
Figure 1. Different NGS-based DNA methylation analysis methods (Jeong et al., 2016).
In terms of the merit and bias of these methods, BS-seq and RRBS can generate a base-resolution DNA methylome, whereas MeDIP-seq can only generate relative enrichment of specific regions across the genome. Chromatin immunoprecipitation (ChIP) offers an advantageous tool for studying the levels of histone methylation associated with a specific gene promoter region between normal and diseased tissues. Identifying the genetic targets of DNA binding proteins and revealing the mechanism of protein-DNA interaction is crucial for understanding cellular processes. Chromatin immunoprecipitation sequencing (ChIP-seq) allows you to make the most of your chromatin studies with minimal sequencing bias.
How to Detect RNA Methylation
Both DNA and RNA methylation involve the enzymatic addition of a methyl group (CH3) to a specific atom on the DNA or RNA molecule, catalyzed by methyltransferases. In cellular RNA, more than 100 types of chemical modifications have already been identified. Among these, there are various types of RNA methylation, including m6A RNA methylation, m5C RNA methylation, m1A RNA methylation, and m7G RNA methylation. Currently, the most prominent and enriched type is m6A RNA methylation, which refers to the methylation of the nitrogen atom at the 6th position of adenine in RNA molecules (N6-methyladenosine, m6A). This modification stands as the most common post-transcriptional modification in eukaryotic mRNA, accounting for 80% of RNA methylation modifications.
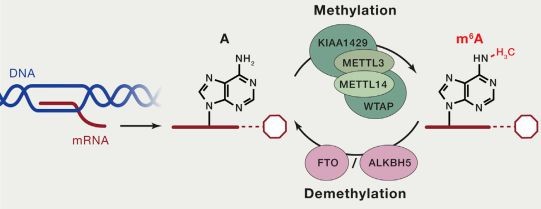
The current methods for detecting RNA methylation primarily include the following:
- MeRIP Sequencing (Methylated RNA Immunoprecipitation Sequencing): This method is based on the principle of antibody-specific binding to methylated bases. It uses m6A-specific antibodies to immunoprecipitate and enrich methylated RNA fragments, which are then subjected to high-throughput sequencing to identify m6A modifications. However, this method can only identify regions with high levels of methylation and cannot achieve single-base resolution for RNA methylation.
- miCLIP (Methylation Individual Nucleotide Resolution Cross-Linking and Immunoprecipitation): In this technique, methylated RNA is specifically bound to antibodies and then cross-linked with ultraviolet light. The reverse transcription of the cross-linked RNA results in cDNA mutations or truncations, indicating the presence of m6A. While miCLIP can identify RNA methylation at single-base resolution, the high costs associated with using isotopic labels such as P32 make it less feasible for routine laboratory use.
- Nanopore Sequencing Technology: This third-generation sequencing technology identifies base sequences based on electrical signals. Different modified bases on RNA cause varying degrees of obstruction as they pass through the nanopore channel, generating characteristic electrical signals. By real-time monitoring of these signals, the corresponding base types and their modifications can be determined. Thus, nanopore sequencing can detect RNA methylation at single-base resolution without the need for specific antibody binding.
Our Epigenomics Sequencing Services
Our DNA methylation solutions include:
Whole Genome Bisulfite Sequencing
Whole Genome Bisulfite Sequencing provides a comprehensive view of DNA methylation patterns across the entire genome, allowing for the examination of methylated and unmethylated cytosines.
Targeted Bisulfite Sequencing
This sequencing methodology selectively captures and examines defined genomic regions, facilitating the precise and cost-effective analysis of DNA methylation patterns.
Reduced Representation Bisulfite Sequencing
Reduced Representation Bisulfite Sequencing is a method that focuses on a subset of the genome, offering a balance between genome coverage and cost-effectiveness for DNA methylation analysis.
MeDIP Sequencing
MeDIP Sequencing facilitates the enrichment and analysis of methylated DNA fragments, offering valuable insights into the patterns of DNA methylation and their potential functional implications.
ChIP-Seq
Chromatin Immunoprecipitation Sequencing (ChIP-Seq) is a powerful method for identifying protein-DNA interactions, elucidating transcription factor binding sites, and exploring epigenetic modifications.
hMeDIP-seq
Hydroxymethylated DNA Immunoprecipitation Sequencing (hMeDIP-Seq) is employed to specifically study DNA hydroxymethylation, a crucial epigenetic mark involved in gene regulation.
ATAC-Seq
ATAC-Seq yields data concerning chromatin accessibility, enabling the identification of both open and closed chromatin regions within the genome.
NGS-BSP
Next-Generation Sequencing of Bisulfite-Converted DNA (NGS-BSP) is a method used for comprehensive DNA methylation analysis, offering single-base resolution insights.
DNA 6mA Sequencing
DNA 6mA Sequencing focuses on the detection and quantification of N6-methyladenine (6mA) in DNA, an emerging epigenetic mark.
DAP-Seq Service
DNA Affinity Purification Sequencing (DAP-Seq) is a method employed to unveil interactions between proteins and DNA, thus enhancing our comprehension of DNA-binding proteins and their functions in gene regulation.
oxBS-seq
Oxidative Bisulfite Sequencing (oxBS-seq) is a specialized method used to discriminate between 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) in DNA, shedding light on their distinct roles in epigenetic regulation.
5mC/5hmC Sequencing
Our high-throughput 5mC/5hmC sequencing service utilizes multiple mature and stable platforms with high efficiency, simplicity, and accuracy to help your epigenetics research.
Our RNA methylation solutions include:
MeRIP Sequencing (m6A Analysis)
MeRIP Sequencing focuses on RNA modifications, particularly N6-methyladenosine (m6A), and aids in understanding post-transcriptional regulation and RNA modification dynamics.
RIP-Seq
RNA Immunoprecipitation Sequencing (RIP-Seq) is employed to explore interactions between RNA and proteins, providing valuable insights into the functions of RNA-binding proteins and their impact on RNA functionality.
2'-O-RNA Methylation Sequencing
The 2'-O-RNA methylation sequencing service (RiboMeth-seq) offers comprehensive detection of 2'-O-RNA methylation modifications across a variety of RNA molecules, including snoRNA, tRNA, and rRNA. RiboMeth-seq primarily utilizes either alkaline or metal ion treatment to induce random RNA cleavage. Positions of 2'-O-methylation exhibit resistance to cleavage, thus remaining intact; conversely, non-methylated sites are prone to cleavage or degradation. By identifying the sites that resist cleavage, RiboMeth-seq effectively maps 2'-O-methylation sites.
Nanopore RNA Methylation Sequencing
Nanopore RNA methylation sequencing is a method that employs nanopore technology to directly sequence RNA molecules and detect their methylation modifications. This technique enables precise localization and identification of methylation sites through real-time analysis of electrical signal changes as RNA molecules pass through the nanopore. With the advantages of direct sequencing and long read lengths, this technology serves as a powerful tool for investigating the roles of RNA methylation in gene expression and cellular function.
Advantages of Epigenomics Sequencing
- Comprehensive Insights: Epigenomics research provides a comprehensive landscape of epigenetic modifications across the entire genome, encompassing DNA methylation, histone modifications, and the regulatory roles of non-coding RNAs. This holistic approach is pivotal for elucidating intricate gene regulatory networks and their dynamic responses.
- Sensitivity and Specificity: Modern epigenomic techniques, such as ChIP-seq, Bisulfite-seq, and ATAC-seq, offer exceptional sensitivity and specificity. These methodologies enable precise detection and quantification of subtle changes in epigenetic marks, essential for deciphering cellular processes and disease mechanisms at a molecular level.
- High-Throughput Analysis: Epigenomic methodologies facilitate high-throughput analysis of thousands of genomic regions and epigenetic loci concurrently. This capability significantly enhances research efficiency, supporting large-scale investigations into biological systems and complex genetic interactions.
- Temporal and Spatial Resolution: Advanced epigenomic tools provide temporal and spatial resolution of epigenetic modifications, capturing dynamic changes across various developmental stages and cell types. This detailed insight into epigenetic dynamics contributes crucially to understanding developmental biology and the pathogenesis of diseases.
Application of Epigenomics Sequencing
- Cancer Research: Epigenomics finds extensive application in cancer research, where analysis of epigenetic alterations in tumor cells identifies potential biomarkers and therapeutic targets. This contributes significantly to advancing personalized treatment strategies.
- Developmental Biology: Studying epigenetic regulatory mechanisms during embryonic development enhances our understanding of cellular differentiation and organogenesis at a molecular level.
- Neuroscience: Exploring the role of epigenetic modifications in neurodevelopment, synaptic plasticity, and neurodegenerative diseases reveals mechanisms underlying neurological disorders.
- Environmental Science: Epigenomics is utilized in studying environmental influences, such as diet, pollutants, and lifestyle, on gene expression and epigenetic modifications. This aids in assessing the relationship between environment and health.
- Plant Science: In plant epigenetic research, epigenomics uncovers molecular mechanisms governing plant growth, development, and responses to environmental stressors, facilitating crop improvement and agricultural productivity.
Sample Requirements
Below are the sample requirements for some of our epigenomic sequencing services. For more detailed information, please refer to the specific service pages. Additionally, if you are interested in our services, please contact us to confirm the sequencing sample requirements.
Table 1. Epigenomics sequencing
| Service | Sample Type | Recommended Quantity | Minimum Quantity | Minimum Concentration |
|---|---|---|---|---|
| MeDIP-Seq/hMeDIP-seq | Genomic DNA | ≥ 2 µg | 1 µg | 20 ng/μL |
| WGBS (Whole Genome Bisulfite Sequencing) |
Genomic DNA Cell Tissue |
≥ 1 μg ≥ 1 x106 ≥ 50 mg |
200 ng |
10 ng/µL |
| RRBS (Reduced representation bisulfite sequencing) |
Genomic DNA Cell Tissue |
≥ 1 μg ≥ 5 x106 ≥ 30 mg |
20 ng 3×103 |
20ng/µL |
| Targeted Bisulfite Sequencing | Genomic DNA Cell Tissue |
≥ 500 ng ≥ 1 x106 ≥ 20 mg |
50 ng | 10 ng/µL |
| oxWGBS-Seq | Genomic DNA | ≥ 3 µg | 1 µg | 30 ng/µL |
| oxRRBS-Seq | Genomic DNA | ≥ 2 µg | 50 ng/μL | |
| oxTBS-Seq | Genomic DNA | ≥ 1 µg | 20 ng/μL | |
| Epityper | Genomic DN | ≥ 1 µg | ||
| DNA 6mA-IP-Seq | Genomic DNA | ≥ 5 µg | 20 ng/μL | |
| ChIP-seq | ChIPed DNA Cell Tissue |
≥ 10 ng ≥ 2 x107 ≥ 500 mg |
5 ng 1×105 |
1 ng/µL |
| DAP-seq | TF Cell Tissue |
≥ 5 µg ≥ 5 x106 ≥ 500 mg |
1 µg 200 mg |
20 ng/µL |
| ATAC-seq | Cell Tissue |
≥ 1 x106 ≥ 500 mg |
5×104 200mg |
1 ng/µL |
Table 2. RNA Epigenomics Sequencing
| Service | Sample Type | Recommended Quantity | Minimum Quantity | Minimum Concentration |
|---|---|---|---|---|
| RIP-seq | IPed RNA Cell Tissue |
≥ 100 ng ≥ 5×107 ≥ 500 mg |
40 ng 200 mg |
5 ng/μL |
| eCLIP-Seq | IPed RNA Cell Tissue |
≥ 100 ng ≥ 3×107 ≥ 500 mg |
40 ng 200 mg |
5 ng/μL |
| MeRIP (Methylated RNA Immunoprecipitation) |
Total RNA Cell Tissue |
≥ 10 μg ≥ 1×107 ≥ 500 mg |
2 μg 200 ng |
1 ng/µL |
| RNA BS-seq (RNA Bisulfite Sequencing) | Total RNA Cell Tissue |
≥ 10 μg ≥ 1×107 ≥ 500 mg |
100 mg |
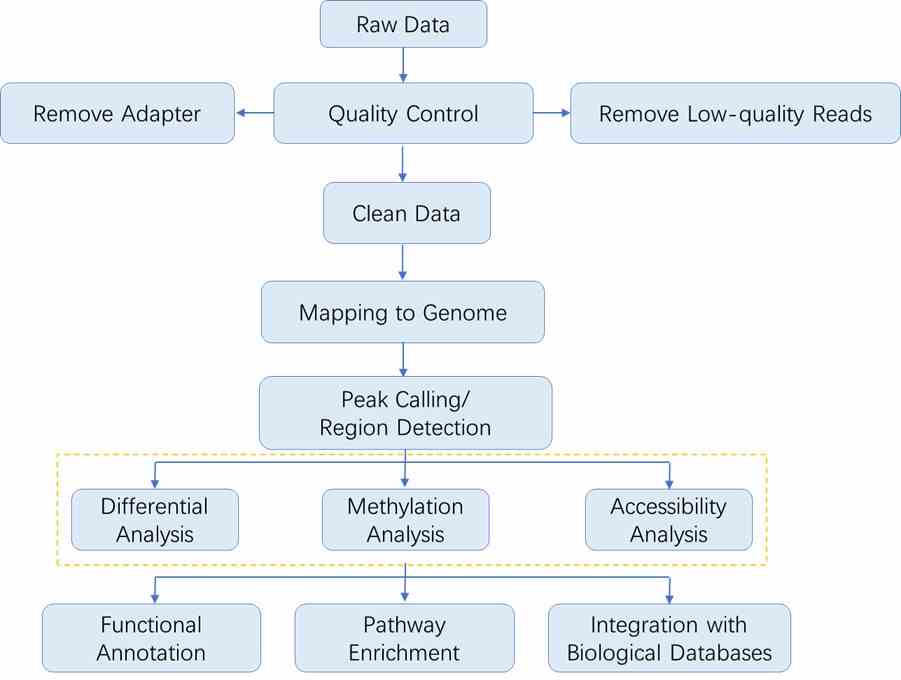
Analysis Pipeline
The sequencing data analysis workflow in epigenomics generally follows the outline depicted below. As each method involves specific steps, you may refer to the respective sections for detailed data analysis procedures.
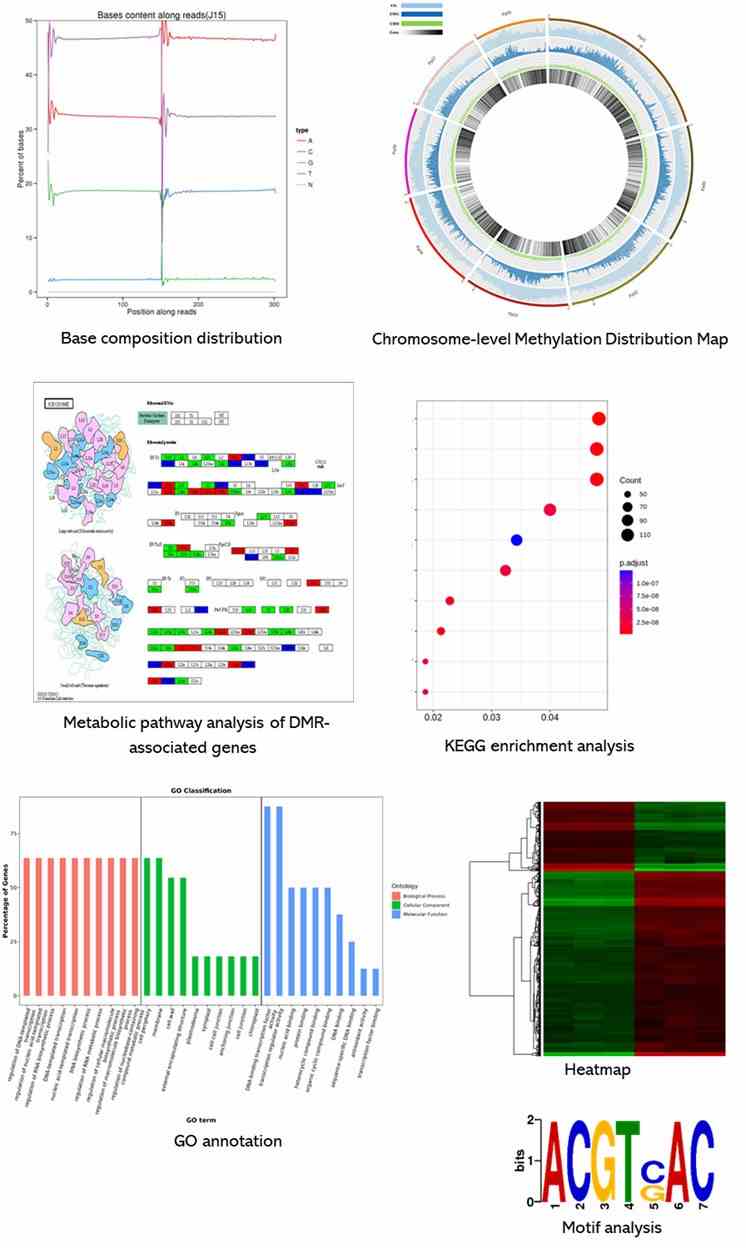
Results Display
Below is a partial presentation of the results from our epigenomic sequencing analyses. For more detailed information, please refer to the specific service pages.
References
- Jeong H.M., et al. Efficiency of methylated DNA immunoprecipitation bisulphite sequencing for whole-genome DNA methylation analysis. Epigenomics. 2016, 8(8):1061-77.
- Roundtree I A, Evans M E, Pan T, et al. Dynamic RNA Modifications in Gene Expression Regulation. Cell, 2017, 169(7):1187-1200.
Disruption of tRNA biogenesis enhances proteostatic resilience, improves later-life health, and promotes longevity
Plos Biology | 2024Methylation in the CHH Context Allows to Predict Recombination in Rice
International Journal of Molecular Sciences | 2022High-Fat Diets Fed during Pregnancy Cause Changes to Pancreatic Tissue DNA Methylation and Protein Expression in the Offspring: A Multi-Omics Approach
International Journal of Molecular Sciences | 2024Folate Carrier Deficiency Drives Differential Methylation and Enhanced Cellular Potency in the Neural Plate Border
Frontiers in Cell and Developmental Biology | 2022