
 Sample Submission Guidelines
Sample Submission Guidelines
Metagenome Sequencing vs. Viral Amplicon Sequencing: Choosing Effective Sequencing Methods for Monitoring Viral Mutations
The monitoring of viral mutations has gained paramount importance due to the likelihood of mutations occurring during viral transmission. This necessitates a meticulous tracking of these mutations, which is compounded by the challenge of enhancing virus detection sensitivity. The crux of the challenge lies in obtaining the entire viral genome sequence to facilitate a comprehensive understanding and monitoring of mutations. Simultaneously, considerations of cost-effectiveness underscore the urgency for numerous entities engaged in testing and research endeavors. In this discourse, we delve into a comprehensive analysis of three robust sequencing methods—Metagenome Sequencing, and Viral Amplicon Sequencing—scrutinizing their applications, advantages, and the intricate considerations involved in monitoring viral mutations.
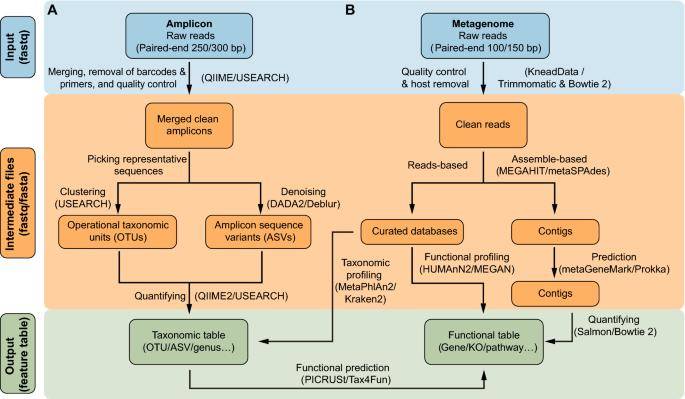 Workflow of commonly used methods. (Liu et al., 2021)
Workflow of commonly used methods. (Liu et al., 2021)
Sequencing Purpose: Rapid Detection or Sequence Assembly in Viral Monitoring
The advent of high-throughput sequencing technology has orchestrated a paradigm shift in our capacity to detect and study novel coronaviruses and other viral pathogens. The decision between rapid detection and meticulous sequence assembly becomes a critical juncture, contingent on the precise aims of the investigation or clinical application, necessitating the judicious selection of appropriate methodologies and strategic approaches.
Rapid Detection: A Swift Response to Emerging Threats
In time-sensitive scenarios, such as disease outbreaks or suspected infections, rapid identification of viral pathogens assumes paramount importance. The arsenal of high-throughput sequencing plays a pivotal role in such contexts. When confronting challenges like confirming diagnoses in patients with negative qPCR test results, discerning complex or secondary infections, or executing extensive screenings of a vast sample pool, high-throughput sequencing emerges as a potent solution.
Advantages:
- Expediency: High-throughput sequencing expedites the identification of viral genetic material, enabling swift responses to potential outbreaks.
- Comprehensive Profiling: This technology enables the concurrent detection of multiple viral species within a solitary sample, facilitating a comprehensive evaluation of the viral landscape.
- Early Alert System: Rapid detection serves as an early alert mechanism, empowering public health authorities to institute timely interventions and containment measures.
Sequence Assembly: Unraveling the Genetic Blueprint
For in-depth and comprehensive analysis, particularly when confronting unknown pathogens or unraveling the genetic makeup of emerging viruses, the sequence assembly approach through high-throughput sequencing becomes indispensable. This methodology entails the acquisition of complete viral genome sequences, allowing researchers to delve into the intricate nuances of viral evolution, mutations, and potential virulence determinants.
Advantages:
- Genetic Elucidation: Sequence assembly offers a granular comprehension of viral genomes, aiding in the identification of pivotal genomic segments and potential targets for drugs or vaccines.
- Evolutionary Insights: High-depth sequencing empowers researchers to trace the evolutionary trajectory of the virus, unmask mutations, and decipher potential patterns of adaptation.
- Epidemiological Insights: Comprehensive genome sequences facilitate thorough epidemiological studies, enabling researchers to trace the virus's propagation and evolution over time.
Methodological Selection
The choice between rapid detection and sequence assembly mandates a multifaceted consideration of various parameters:
- Urgency: The urgency of the situation and the imperative for immediate diagnostic insights.
- Resources: The availability of laboratory resources encompassing equipment, personnel, and computational capabilities.
- Research Goals: The specific objectives of the research, be it epidemiological studies, vaccine development, or the pursuit of insights into viral evolution.
- Sample Availability: The quantity and quality of available samples for sequencing.
Sequencing Approaches: Metagenome Sequencing vs. Targeted Sequencing
In the realm of high-throughput sequencing for investigating viral genomes, two distinct methodologies emerge as potent tools: Metagenome sequencing and targeted sequencing. Each approach boasts unique strengths, shaping their suitability based on specific applications and research objectives.
| Aspect | Metagenome Sequencing | Targeted Sequencing |
| Approach | Comprehensive and unbiased exploration of microbial genomes including bacteria, viruses, and other microorganisms | Precision-oriented focus on specific viral genomes or target microorganisms |
| Advantages | - Unbiased discovery of novel pathogens | - Precision enrichment and detailed sequencing of specific viral genomes or target microorganisms |
| - Broad spectrum detection of pathogenic microorganisms | - Cost-efficient and suitable for projects with limited resources | |
| - High sensitivity for detecting low viral loads and genetic variations | ||
| Considerations | - Generates substantial data volume requiring robust computational resources | - Focus limited to predefined viral genomes or target microorganisms |
| - Not ideal for identifying unknown pathogens | ||
| - Comprehensive approach may include data on non-target microorganisms | - Specialized for specific virus detection or detailed analysis of well-characterized pathogens | |
| Recommendations Based on Application | Optimal for unexplained infections, mixed infections, and discovery of unknown pathogens. Provides comprehensive insights. | Meticulous examination of specific viral genomes or known microorganisms |
Data Volume in Sequencing: Tailoring Quantity to Method and Objective
The magnitude of sequencing data bears pivotal relevance in the success of viral genome analysis, dictated by the intended sequencing purpose, chosen methodology, viral load, and other pertinent factors. Striking an equilibrium between data volume and specific requisites assumes critical importance in achieving precise outcomes and meaningful insights. Broadly, an increased data volume augments sensitivity, elevates clinical test positivity rates, and fortifies the effectiveness of viral sequence assembly. Vital considerations encompass attaining viral genome coverage exceeding 95% and a single base depth of at least 10x.
Metagenome Sequencing: Adapting Data Volume to Sample Attributes
For metagenome sequencing, the optimal data volume hinges on the nature of the sample material under scrutiny.
- Samples with Viral Loads >104 copies/ml or qPCR CT Values <24.5:
- Recommended Data Volume: 20Gb (PE100, 100M reads)
- Rationale: Amplifying data volume in such cases enhances sensitivity and increases the likelihood of yielding positive clinical test results.
Amplicon Sequencing for Samples with Very Low Viral Load
Metagenome sequencing becomes less effective when confronted with samples bearing very low viral loads, especially those exhibiting CT values surpassing 28.7. In such instances, an alternative approach, such as multiplex PCR amplicon sequencing, is advisable for achieving precise and accurate results.
Multiplex PCR Amplicon Sequencing: Precision in the Presence of Low Viral Load
When deploying multiplex PCR amplicon sequencing, the optimal data volume ranges from 5 to 20 million reads. This technique proves exceptionally valuable when scrutinizing samples with exceedingly low viral loads, including those housing fewer than 102 copies/ml.
Sequencing Library Processing for Viral RNA Sequencing
Within the realm of viral RNA sequencing, the determination of whether to eliminate ribosomal RNA (rRNA) during library preparation emerges as a pivotal deliberation that profoundly impacts the quality and success of the sequencing process. While rRNA constitutes a significant fraction of total RNA (often exceeding 80%), its removal can enhance the efficiency and utility of generated sequencing data. Nonetheless, the decision to remove rRNA is intricate, contingent on diverse factors, including sample characteristics, input material, and library construction objectives.
The Significance of rRNA Removal
Ribosomal RNA assumes a dominant role in cellular RNA, serving as a fundamental actor in protein synthesis. In the context of viral RNA sequencing, the presence of rRNA can dilute the desired viral RNA content, potentially hampering the detection and faithful representation of viral sequences. Through selective rRNA removal, researchers can heighten the sensitivity of viral detection, improve library construction efficiency, and ultimately amplify the pertinence of sequencing data for viral analysis.
Sample Considerations
- Limited Viral Nucleic Acid Input: In scenarios featuring scarce viral nucleic acid input, such as in low viral load situations, rRNA removal could prove particularly advantageous. Minimizing rRNA interference enables focused capture of viral sequences, conceivably yielding more precise and comprehensive outcomes.
- Degraded Samples: Samples subject to prolonged storage and significant degradation might harbor meager quantities of intact viral RNA. In such cases, rRNA removal might exacerbate the challenges of library preparation by further diminishing available RNA material. In these scenarios, researchers might opt against rRNA removal to maximize the likelihood of successful library construction.
Balancing rRNA Removal and Library Construction
While rRNA removal offers benefits in numerous cases, achieving equilibrium that aligns with library construction goals and sample attributes is crucial. In certain scenarios, optimizing library preparation success might supersede rRNA removal, particularly when confronted with samples marked by low viral abundance or substantial degradation.
Summary
- For comprehensive analysis of all potential pathogenic microorganisms, including the target microorganism, in samples with a viral load ≥105 copies/ml or a Ct value ≤ 24.5, macro-genomic sequencing is the recommended approach.
- When focusing solely on the target microorganisms and dealing with challenging samples (viral load <105 copies/ml or Ct >24.5), probe capture sequencing is preferable for detecting minor base mutations and frequencies, while multiplex PCR amplicon sequencing is advised for capturing major base mutations.
- For targeted analysis of the specific microorganism, prioritizing major base mutations, and working with difficult samples (viral loads as low as 102 copies/ml or Ct values as high as 35), multiplex PCR amplicon sequencing is the recommended method.
Reference:
- Liu, Yong-Xin, et al. "A practical guide to amplicon and metagenomic analysis of microbiome data." Protein & cell 12.5 (2021): 315-330.


