
 Sample Submission Guidelines
Sample Submission Guidelines
Identification of Disease-Related Cell States by Single-Cell Sequencing
In recent years, technological breakthroughs in single-cell and spatial genomics have opened new avenues for cellular and tissue mapping studies in health and disease. The precise identification of cellular states in disease using single-cell genomics can provide insight into pathogenesis, biomarkers, and potential drug targets. The standard approach for identifying cellular states involves the combined analysis of single-cell RNA sequencing (scRNA-seq) data from diseased and healthy reference tissues. Of these, the selection of healthy reference datasets is critical.
Currently, the Human Cell Atlas Consortium has performed large-scale analyses of healthy samples and published large-scale, unified datasets or atlas datasets from multiple organs, but the characteristics of samples included in this atlas may be very different from those of samples in disease cohorts. Collecting control samples from healthy tissues that match disease samples with similar demographic and clinical characteristics can greatly reduce information errors due to confounding factors.
The research team evaluated how the choice of single-cell reference atlas affects the identification of cell state changes from disease sample scRNA-seq data. Using publicly available single-cell transcriptome data, a single-cell reference atlas from healthy individuals was created, confirming that using the atlas for latent space (LST) learning and difference-in-difference analysis of matched controls better identifies disease-associated cells, particularly multiple disturbed cell types. This study provides guidance for designing disease cohort studies and optimizing the use of cellular atlases.
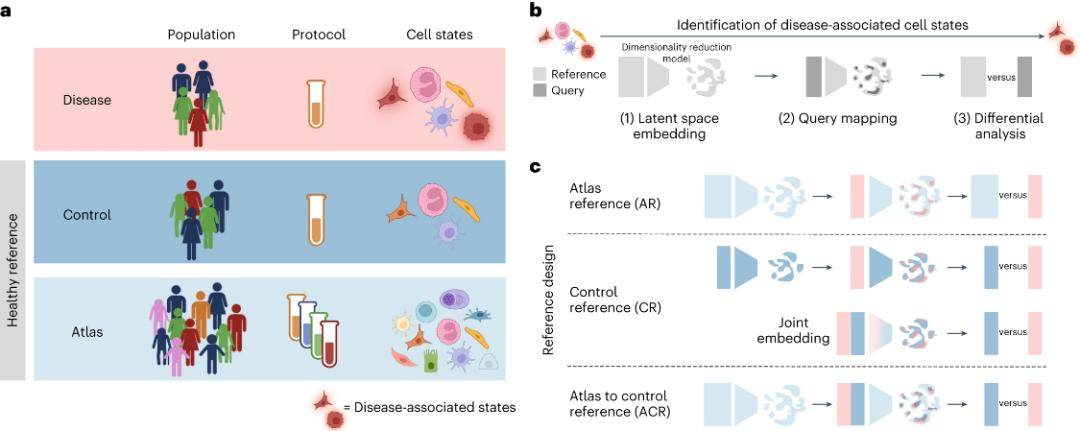 Using healthy reference datasets to discover disease-associated cell states. (Dann et al., 2023)
Using healthy reference datasets to discover disease-associated cell states. (Dann et al., 2023)
Interferon (IFN) in COVID-19 Patients Detection of Stimulation Status
The research team used publicly available scRNA-seq datasets from 90 COVID-19 patients and 23 healthy donor PBMCs; and selected scRNA-seq profiles from 1,219 healthy individuals from 12 studies as the mapping dataset; potentially embedded using healthy PBMC profiles (ACR design) was compared to a mapping dataset using only COVID-19 and a control dataset with a combined embedding (CR design) were compared to a control dataset. To quantify the ability of the different designs to identify disease-associated states, the team examined cells expressing genes related to the interferon (IFN) signaling pathway, a key antiviral response pathway and a recognized marker for COVID-19.
By integrating data from the COVID-19 cohort and the PBMC cell atlas, the team found that utilizing an ACR design allowed for more sensitive identification of transitional and heterogeneous pathological cell states. Within the COVID-19 dataset, the team captured the IFNhi status of different immune cell types, as well as subdivided subpopulations of dysfunctional CD14+ monocytes that correlated with disease severity.
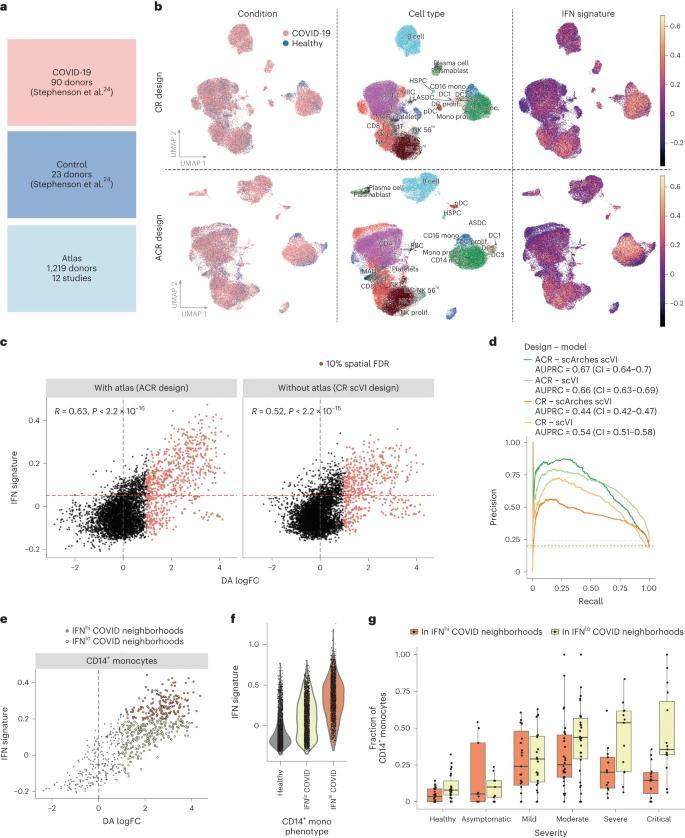 Detection of cell states associated with COVID-19 in a case-control cohort with a healthy atlas. (Dann et al., 2023)
Detection of cell states associated with COVID-19 in a case-control cohort with a healthy atlas. (Dann et al., 2023)
Detection of Abnormal Cell Status in Pulmonary Fibrosis
To assess the benefits of using single-cell profiles and control datasets jointly in other biological contexts, the research team analyzed scRNA-seq datasets from lung tissue samples from 32 patients with idiopathic pulmonary fibrosis (IPF) and investigated the disease state of pulmonary fibrosis using single-cell profiles of healthy lung tissue. The study included data from 28 control donors and 18 chronic obstructive pulmonary disease (COPD) patients, and the core Human Lung Cell Atlas (HLCA) dataset was selected as the atlas dataset.
The research team used an ACR design to analyze the data from the IPF patients and identified two rare abnormal basal cell states associated with the disease, KRT5-KRT17+ basal cells and KRT5+KRT17hi basal cells. The above cells were characterized separately to further identify differentially expressed genes (DEGs) that were differentially expressed in the abnormal basal-like state and overexpressed compared to normal basal cells. A total of 981 significant DEGs were identified, including 6 KRT17hi abnormal basal cell markers and 35 basal cell markers previously described, enhancing the understanding of the basal-like cell phenotype in IPF.
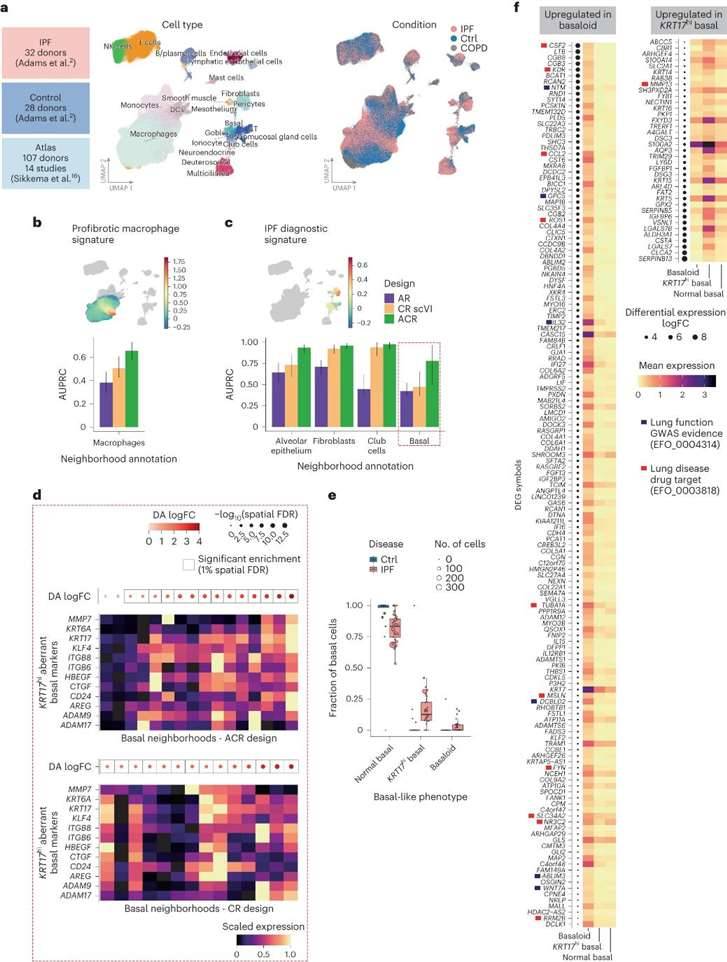 Detection of cell states associated with IPF. (Dann et al., 2023)
Detection of cell states associated with IPF. (Dann et al., 2023)
Reference:
- Dann, Emma, et al. "Precise identification of cell states altered in disease using healthy single-cell references." Nature Genetics (2023): 1-11.


