
 Sample Submission Guidelines
Sample Submission Guidelines
How Can Sequencing Tap into Microbial Associations with Diseases?
The human microbiome, as the second largest human genome, is potentially associated with many diseases. The study of microbe-associated diseases has become a key focus in the field of medicine, providing new explanatory directions for diseases with unknown pathologic mechanisms. However, as microbial communities are susceptible to environmental changes, individual differences lead to significant differences in human microbial communities. Therefore, in the field of medical microbiology, the results from a small number of samples may be subject to greater instability.
Bulk Microbiome (BM) has become the focus of microbial-disease association research with its more stable results of big data and ultra-excellent cost-effectiveness of low cost and high output. However, microbiology is a relatively unfamiliar field for many medical researchers, and this, coupled with the amount of large sample data, is often daunting. So, how should we tap into the potential association between microbes and diseases through the idea of large-sample microbiome construction analysis?
Biomarker analysis programs are usually implemented in conjunction with population analysis programs. First, by determining whether there is a clear distinction between the groups, if there is a clear difference between the groups, then we can further dig into the biomarkers that cause such a significant difference. If the groups are not clearly differentiated, the biomarker information causing the phenotypic differences can be mined deeper based on the original grouping, and the potential role of microorganisms on the differential phenotypes can be discussed. In addition, validation of the mined biomarker by other means can improve the confidence of the results.
Case Study: Urologic Microbiome
For example, the diagnostic complexity of urinary tract infections (UTIs) lies in the fact that symptomatic patients with a lack of traditional urinary pathogens or bacteria are often encountered in traditional urine culture tests. As a result, there is a lack of definition of a healthy urinary microbiome. A comprehensive survey of the urinary microbiomes of patients with UTIs and healthy populations through large-scale metagenomic sequencing will help determine the role of the microbiome in practical medical testing.
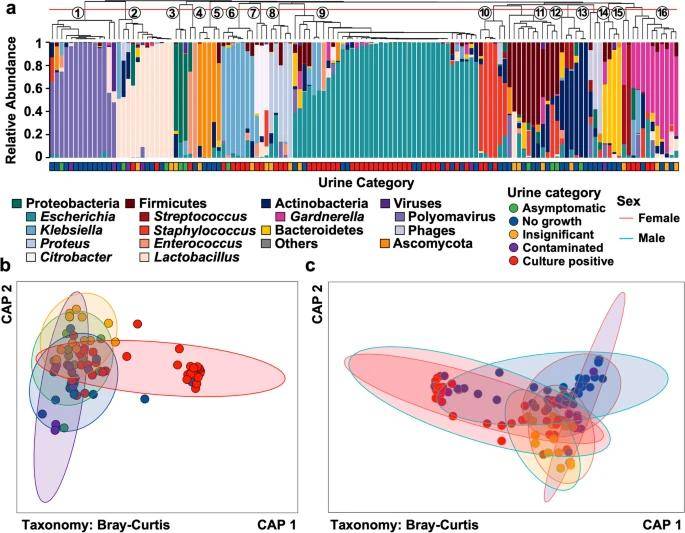 Microbiota composition varies between categories of diagnostic urinanalysis. (Adu-Oppong et al., 2022)
Microbiota composition varies between categories of diagnostic urinanalysis. (Adu-Oppong et al., 2022)
Patients were categorized into four groups based on standard urine culture types:
(1) Culture positive: significant growth of one or two uropathogenic bacteria;
(2) Contaminated: growth of three or more bacteria at concentrations higher than 105 colony-forming units/mL;
(3) Insignificant: the culture process was lower than 105 colony forming units/mL;
(4) No-growth group: the culture process showed no obvious signs of microbial growth.
At the same time, healthy individuals were selected as a control group for sampling and two additional samples were taken to track changes in their urinary microbial dynamics.
This article utilizes a combination of population and marker analysis protocols. Population analysis explores differences in urinary microbial composition between healthy individuals and patients with UTIs and the dynamics of urinary microbial changes in healthy individuals. Marker analysis, on the other hand, mined the differential microbial composition between patients with UTIs and healthy individuals and identified the differences in microbial composition by comparing sequencing identifications with standard culture. Through large-sample metagenomic sequencing, this paper mined the differences in urologic microbial phenotypes between patients with UTIs and healthy individuals, and compared the microbial differences between traditional culture identification and sequencing identification.
The key contribution of this study is the use of large sample data analysis to demonstrate the relationship between disease phenotypes and microbial composition, with broader coverage and more stable results. Also, the study was made more convincing by statistically analyzing the similarity of microbial composition between healthy individuals and patients with UTIs. The results of this study break the traditional perception of UTIs and confirm that the disease is not strongly associated with microbial phenotype. The large-sample microbiome played a key role in this process, firstly by emphasizing the impact of individual dynamic differences on the results through the wider coverage of the large-sample data, and secondly by being more convincing on a statistical level.
Through the combination of population and marker analyses, this paper provides a powerful methodology for exploring microbial-disease associations, especially in the context of coping with large sample data. This approach has not only achieved significant results in UTIs studies, but also provides useful lessons for future medical microbiome research.
Reference:
- Adu-Oppong, B., Thänert, R., Wallace, M.A. et al. Substantial overlap between symptomatic and asymptomatic genitourinary microbiota states. Microbiome 10, 6 (2022).


