
 Sample Submission Guidelines
Sample Submission Guidelines
Integrated Analysis of Microbiome and Transcriptome Data Unraveling the Dynamic Interplay between Host and Microbial Communities
In the quest to comprehend the intricate interplay between organisms and their environment, researchers have increasingly turned their attention to the joint investigation of transcriptomes and microbiomes. This cutting-edge approach seeks to shed light on the dynamic relationships between host organisms and the diverse microbial communities residing within them. By integrating transcriptomics and microbiome analysis, scientists are now able to explore the molecular mechanisms underlying these interactions, leading to groundbreaking discoveries in various fields, from health and disease to ecology and beyond.
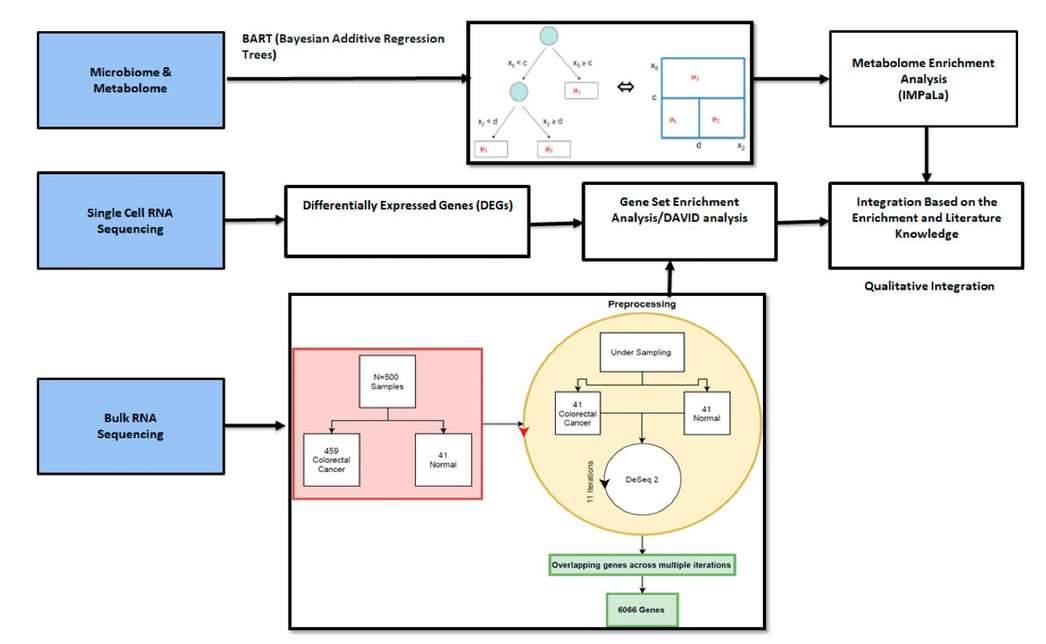 Integration of different methods and omics data sets. (Bisht ET AL., 2021)
Integration of different methods and omics data sets. (Bisht ET AL., 2021)
How To Conduct Joint Analysis on Transcriptome & Microbiome?
Conducting joint research on the transcriptome and microbiome involves investigating the intrinsic regulatory mechanisms of the phenotype from both the host gene and bacterial community perspectives. The transcriptome primarily targets differentially expressed genes and functional pathways at the host gene level, while the microbiome focuses on analyzing the impact of changes in the host phenotype/health status on bacterial community balance and vice versa. By combining these approaches, the relationship between the host phenotype and the microbiome can be explored comprehensively.
Attention Points for Joint Transcriptome & Microbiome Research:
- Sample Selection: Choose appropriate samples based on research objectives. For microbiome research, consider microbial diversity, leading to more extensive sample selection. For macro-genome research, opt for samples with lower host content to minimize host interference and ensure data quality.
- Biological Replicates: To obtain reliable results, use a sufficient number of samples in both transcriptome and microbiome analyses. Aim for at least 3-5 samples per group for each analysis. If conducting histological association analysis, maintain a one-to-one correspondence between transcriptome and microbiome samples, ensuring the same number of samples in each group (≥30 cases/group for clinical samples and ≥15 cases/group for WGCNA analysis).
- Sample Preservation: If using the same sample for both transcriptome and microbiome studies, divide the sample into two parts and store them at -80°C. For valuable samples, it is advisable to keep a backup sample for future reference.
In the joint research, it is also crucial to correlate differentially expressed genes/functional pathways with specific bacterial flora to identify key microorganisms that regulate host gene expression or key host genes influencing the balance of key flora. To establish causal relationships between host genes and flora, experiments such as Fecal Microbiota Transplantation (FMT) can be conducted. By following these guidelines, researchers can gain valuable insights into the complex interactions between the transcriptome, microbiome, and host phenotype/health status.
Investigating Neurobehavioral Toxicity in Fish Induced by Micro/Nanoplastics
The neurobehavioral toxicity induced by micro/nanoplastics in the fish, Ctenopharyngodon idellus, was explored from the perspective of the "gut-brain axis." Microbial diversity sequencing of the fish intestine revealed alterations in potential neurotransmitter-secreting species, with increased abundance of Clostridia and Bacillus in the presence of nanoparticles (NPs) and decreased abundance in the presence of microfibers (MFs). Transcriptome sequencing of fish brain tissue identified enriched pathways related to neuroactive ligand-receptor interactions and 5-hydroxytryptaminergic synapses in both NPs and MFs groups, while dopaminergic synaptic pathways were only enriched in the MFs group. These findings suggest a novel neurotransmitter mechanism by which micro/nanoplastics may induce behavioral toxicity through the brain-gut-microbiota axis, without involving joint histological analysis.
Comparative Analysis of Root Transcriptomes and Microbiomes in Salinity-Tolerant Wheat
Researchers presents a comprehensive comparison of the root transcriptomes and microbiomes between the salinity-tolerant wheat introgression line SR4 (derived from a somatic cross between wheat and tall fescue) and its parental wheat variety JN177. The study aimed to understand the mechanisms of gene expression regulation related to root microbial architecture in JN177 and SR4. Through joint analysis of transcriptome and microbial diversity data, co-expression modules in normal and saline soils were identified using weighted gene co-expression network analysis (WGCNA). Hub genes within these modules were also characterized. Additionally, the article investigated the relationship between characteristic gene values of the modules and different enriched microbial taxa. Moreover, genes that regulate the wheat root microbiome under saline stress were further identified.
Microbial Diversity and Transcriptome Analysis in Colonic Mucosa of CF Patients
Researchers explored changes in gene expression and flora composition in the colonic mucosa of patients with cystic fibrosis (CF) using microbial diversity and transcriptome sequencing. By analyzing the host-microbe interactions, the study suggested that these interactions may play a crucial role in the pathogenesis of CF colorectal cancer. The investigation utilized correlation heatmap, correlation network diagram, and other methods to understand the associations between gene expression and microbial composition. These findings provide potential biomarkers for predicting colorectal cancer risk in CF patients.
References:
- Bisht, Vartika, et al. "Integration of the microbiome, metabolome and transcriptomics data identified novel metabolic pathway regulation in colorectal cancer." International journal of molecular sciences 22.11 (2021): 5763.
- Fyhrquist, Nanna, et al. "Microbe-host interplay in atopic dermatitis and psoriasis." Nature communications 10.1 (2019): 4703.
- Hou, Shiji, et al. "A microbiota–root–shoot circuit favours Arabidopsis growth over defence under suboptimal light." Nature Plants 7.8 (2021): 1078-1092.


