
 Sample Submission Guidelines
Sample Submission Guidelines
Advancing Cancer Research through Whole Exome Sequencing
While exons constitute a mere 1-2% of the human genome, they harbor a staggering 85% of known disease-causing mutations, with ongoing exploration into undiscovered disease-related mutations. Emerging as a powerful tool, Whole Exome Sequencing (WES) zeroes in on these exonic regions and neighboring genetic segments. It employs second-generation sequencing to unravel genetic sequences, fusing findings with exon databases and biobanking techniques to illuminate links between mutations and diseases. Remarkably, WES achieves sequencing depths exceeding 100X, rendering it an extraordinarily efficient avenue for unearthing insights into human ailments.
At present, WES technology has permeated domains spanning single-gene disorders, complex maladies, and notably, cancer research—a domain of paramount complexity and significance. Among these pursuits, cancer research stands out as a fervent endeavor, grappling with unrivaled intricacy and universal urgency. CD Genomics stands as a torchbearer, providing pioneering sequencing and biosignature analysis services tailored for the study of tumor exons via WES and biosignature methodologies. This ensures a more efficient, comprehensive, and profound approach to tumor-focused investigations.
Illuminating Mutation Characteristics
Cancer's evolutionary journey is delineated into three pivotal stages—pre-cancerous development, cancer genesis, and tumor differentiation. Throughout these stages, mutations accumulate in diverse patterns, engendering distinct mutational frequencies at individual bases. This phenomenon births unique mutation profiles for each stage. Investigating these distinctive features promises insights into tumor development modes—linear or branching—furnishing invaluable guidance for therapeutic interventions.
The landscape of mutation characterization traverses four categories according to mutation types: Single Base Substitution (SBS), Double Base Substitution (DBS), Small Fragment Insertions and Deletions (ID), and Large Fragment Insertions and Deletions (CN). SBS encapsulates mutations in a single base, totaling 96 mutation types upon permutation and combination analysis. The COSMIC database, a repository of cancer mutations, boasts 78 SBS tumor genome mutation signatures, exemplifying the magnitude of genetic variation within cancer cells.
Furthermore, mutation characterization proves instrumental in examining limited samples like organoids (PDOs). Researchers engineer a gastric cancer organoid for drug testing. Before such tests, confirming organoid alignment with primary tumor traits becomes paramount. WES evaluates two tumor samples, juxtaposing primary tumor and organoid mutation profiles. Remarkably, while general trends in SBS mutation types align, the conclusive correspondence demands supplementary analyses, underscoring the painstaking steps undertaken to verify PDO-primary tumor consistency.
Tumor Mutational Burden Analysis (TMB)
Tumor Mutational Burden (TMB), a metric denoting the count of somatic mutations per megabase (MB) of the longest transcript sequence, undergoes fluctuations across different tumors. This analytical dimension holds the promise of revolutionizing predictive precision in immunotherapy outcomes and stands as a potent catalyst in broadening the landscape of eligible patients for immune checkpoint inhibitor (ICI) therapies. Notably, studies spotlight its pivotal role in predicting immunotherapeutic efficacy within lung, liver, colorectal, bladder, and melanoma cancers. Elevated TMB could stimulate the generation of an abundance of neoantigens, intensifying the likelihood of T-cell recognition—a crucial factor in rendering tumors amenable to immunotherapy. This underscores its pivotal clinical correlation with immune checkpoint inhibitors (ICIs).
Yet, the current scientific panorama reveals that although TMB stands as a promising immunotherapy biomarker in solid tumors, a unanimous consensus on the optimal TMB threshold for distinct solid malignancies remains elusive. This intricate puzzle gains further nuance with the recognition that finely delineating the ideal TMB thresholds holds the potential to orchestrate a dramatic metamorphosis in the efficacy of immune checkpoint inhibitor (ICI) treatments, particularly for non-small cell lung cancer (NSCLC) patients.
Take, for instance, the work of Biagio Ricciuti et al, who delved into the intricate correlation between heightened TMB levels and the efficacy of immunotherapy within the realm of clinically programmed cell death (PD-1/PD-L1) interactions in NSCLC patients. Their inquiry showcased that, in contrast to the TMB-low cohort, the TMB-high group among the PD-1/PD-L1-expressing subset treated with PD-1/PD-L1 inhibition experienced a notable surge in both effective rates and survival outcomes. The hallmark of this augmentation was accompanied by heightened infiltration of CD8+ T-cells, accompanied by a distinctive gene expression profile indicative of an active immune response.
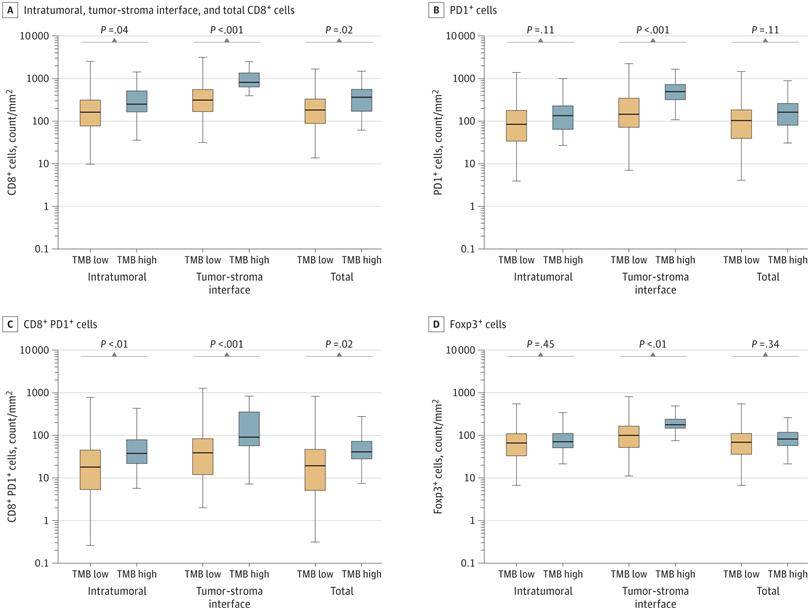 Data are from patients in the Dana-Farber Cancer Institute cohort, including 384 patients with low tumor mutation burden
Data are from patients in the Dana-Farber Cancer Institute cohort, including 384 patients with low tumor mutation burden
(TMB) and 44 patients with high TMB. (Ricciuti et al., 2022)
Loss of Heterozygosity (LOH) through WES Analysis
Loss of heterozygosity (LOH) encapsulates a profound phenomenon where genomic loci that traditionally exhibit heterozygosity in normal tissues undergo a transformation within tumor tissues. This transformation involves the conversion of some heterozygous loci to a state of homogeneity. Mechanisms underpinning this shift include alterations in chromosome copy numbers, gene substitutions, somatic cell recombination, and mitotic nondisjunction. The outcome of this transformative process is termed heterozygous deletion—a pivotal occurrence with a strong affiliation to tumor suppressor genes (e.g., TP53). This deletion curtails the potential for malignant tumor development when both alleles are intact. However, when one allele undergoes substantial aberration, the other allele succumbs to an inactive state due to deletion, erasing its inhibitory role. Subsequently, cellular transformation into a cancerous state ensues.
The immense potential of Whole Exome Sequencing (WES) shines brilliantly in the context of LOH analysis. By leveraging WES data, one can discern regions within the tumor displaying LOH. Simultaneously, the approach uncovers Single Nucleotide Variation (SNV) mutation sites within these LOH regions while furnishing essential annotations about oncogenic attributes. A comprehensive exploration of LOH not only unearths novel LOH-driven oncogenes but also casts a radiant light on the intricate landscape of tumorigenesis.
Beyond its impact as a comprehensive marker, LOH operates as an intricate translator, conveying LOH events within cancer cells into potent activation signals for immune cells. This very facet opens doors for innovative avenues in immunotherapy. The cues derived from LOH could potentially guide immunotherapeutic strategies, training the spotlight on these pivotal events and rendering them susceptible to targeted immune interventions.
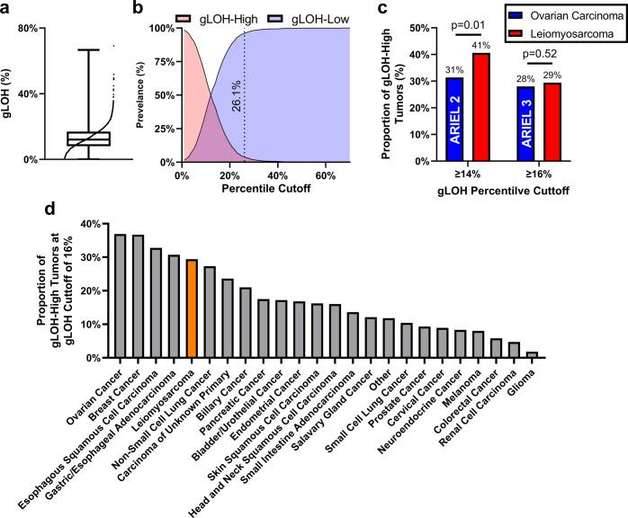 Distribution of gLOH in LMS. (Seligson et al., 2022)
Distribution of gLOH in LMS. (Seligson et al., 2022)
References:
- Ricciuti, Biagio, et al. "Association of high tumor mutation burden in non–small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels." JAMA oncology 8.8 (2022): 1160-1168.
- Seligson, Nathan D., et al. "Drivers of genomic loss of heterozygosity in leiomyosarcoma are distinct from carcinomas." NPJ Precision Oncology 6.1 (2022): 29.


