
 Sample Submission Guidelines
Sample Submission Guidelines
Application of RAD Sequencing Technology
Owing to its potent capacity for detecting genomic variations comprehensively, RAD sequencing technology has found utility in more than 20 animal and plant species that lack a reference genome. Its applications encompass the establishment of genomic markers, genetic and comparative mapping, high-resolution mapping of trait-associated genes and QTLs (Quantitative Trait Loci), investigations in population genetics and evolution, as well as comprehensive genome-wide association studies. Additionally, for species equipped with reference genomes, particularly those boasting larger genomes, RAD sequencing confers a substantial reduction in sequencing costs when compared to whole-genome resequencing. This advantage becomes particularly conspicuous when substantial sample sizes are essential for population genetics investigations.
Development of Molecular Markers
In species without a reference genome, the process of obtaining molecular markers typically involves searching for polymorphisms using previously published markers from the same species or closely related species. Alternatively, the sequence information of these markers and the conserved gene sequences from closely related, sequenced species can be leveraged for marker development. Markers generated using these approaches often consist of SSR markers with limited polymorphism, necessitating the design of numerous primers to yield a small number of polymorphic markers.
In contrast, RAD sequencing technology provides notable advantages for marker development by enabling the discovery of numerous SNPs throughout the genome via sequencing. When compared to SSR markers, SNPs are densely distributed within the genome. Furthermore, RAD sequencing is both time and cost-efficient, resulting in significant reductions in labor and resource expenditures.
Genetic map of wheat with different marker
| Aspect | Sanger Sequencing | Whole Plasmid Sequencing (NGS) |
| Methodology | Chain-termination method | Utilizes Next-Generation Sequencing (NGS)or Long Read Sequencing technologies |
| Sequence Length | Suitable for shorter fragments (up to 1,000 bp) | Can sequence entire plasmids, regardless of size or complexity |
| Speed | Sanger Sequencing can involve extended processing times and is particularly suited for small-scale projects. | In contrast, Whole Plasmid Sequencing using NGS technologies provides a swifter turnaround, making it well-suited for high-throughput applications. |
| Cost | Can be cost-prohibitive for larger plasmids due to multiple sequencing reactions | Generally more cost-effective, especially for larger plasmids, as it involves fewer sequencing reactions |
| Accuracy | Sanger Sequencing is renowned for its exceptional precision and minimal error rates. | Whole Plasmid Sequencing maintains a high degree of accuracy and incorporates enhanced error correction methods. |
| Applicability | Sanger Sequencing is appropriate for smaller plasmids but may encounter challenges when dealing with larger, intricate, or repetitive plasmid structures. | Whole Plasmid Sequencing is highly versatile and can accommodate plasmids of various sizes and complexities, rendering it suitable for comprehensive plasmid analysis. |
Genetic and Comparative Map Construction
Traditionally, the establishment of whole-genome genetic maps using PCR markers was a process characterized by labor-intensive, resource-intensive, and time-consuming aspects, primarily due to the necessity for individual genotyping of each marker within segregating populations through PCR electrophoresis. However, the adoption of RAD sequencing technology for the parents and their segregating populations has revolutionized this practice, enabling the rapid acquisition of a large number of SNP genotypes that offer extensive coverage of the entire genome for the construction of genetic maps.
Genetic maps with heightened marker density and coverage provide a more abundant array of loci and regions for comparative genomics analysis. Consequently, this approach facilitates a more comprehensive and detailed comprehension of the genome and macro-level insights into collinearity relationships when compared to closely related species with known sequences. Notably, this method offers distinct advantages for the detection and characterization of structural variations such as inversions, deletions, and translocations.
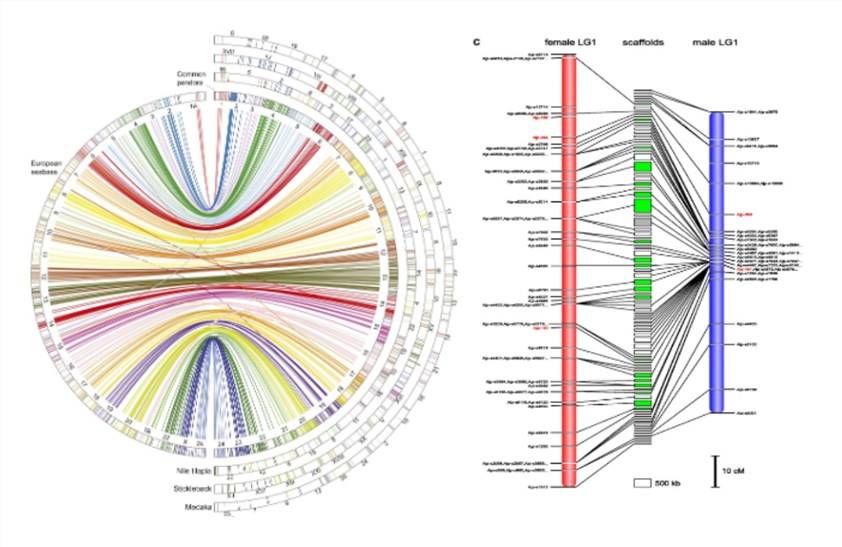
High-Resolution Trait Gene/QTL Mapping
Botanists and genetic breeding researchers are keenly interested in deciphering the link between genotype and phenotype. Uncovering the genes/QTLs (Quantitative Trait Loci) that govern target traits requires high-resolution genetic mapping. The higher the precision of the mapping, the more advantageous it becomes not only for subsequent gene/QTL cloning but also for marker-assisted breeding of target genes.
The initial mapping of genes/QTLs necessitates screening across the entire genome. For qualitative trait genes, Bulked Segregant Analysis (BSA) is commonly employed, whereas the preliminary mapping of QTLs requires scanning the genome using a comprehensive genetic map. Traditional genetic maps constructed using PCR markers vary in marker density across the genome. If a target gene happens to reside in a region with low marker density (e.g., no markers within approximately 30 cM genetic distance), it may be challenging to obtain linked markers through PCR selection. Insufficient marker density similarly affects the accuracy of QTL mapping.
Beyond mapping accuracy, the SNP sequence data derived from RAD sequencing is amenable to comparative analysis with the genome sequences of closely related species. In cases where the SNPs within the mapped region are situated within the coding regions of genes of interest, statistical analyses can be employed to assess these SNP variants. This analysis can unveil which genes have undergone non-synonymous substitutions or premature termination between the parental strains. Such insights provide a valuable reference for the prediction of candidate genes and can streamline subsequent validations of gene function.
Population Genetic Analysis and GWAS
The application of RAD sequencing technology in population genetic analysis and Genome-Wide Association Studies (GWAS) primarily involves sequencing and variant identification in natural populations of the target species. It includes statistical analysis of variant sites within these populations, investigating population structure, and identifying loci in the genome subject to selection.
Concurrently, in combination with phenotype data, it identifies SNPs closely associated with target traits. Subsequent joint analysis of selected loci and SNPs obtained from GWAS facilitates the exploration of the relationship between artificial selection and population evolution.
References:
- Kai W, Nomura K, Fujiwara A, et al. A ddRAD-based genetic map and its integration with the genome assembly of Japanese eel (Anguilla japonica) provides insights into genome evolution after the teleost-specific genome duplication. BMC genomics, 2014, 15(1): 233.
- Kundu A, Chakraborty A, Mandal N A, et al. A restriction-site-associated DNA (RAD) linkage map, comparative genomics and identification of QTL for histological fibre content coincident with those for retted bastfibre yield and its major components in jute (Corchorusolitorius L., Malvaceaesl). Molecular breeding, 2015, 35(1): 19.
- Manousaki T, Tsakogiannis A, Taggart J B, et al. Exploring a nonmodel teleost genome through rad sequencing—linkage mapping in Common Pandora, Pagellus erythrinus and comparative genomic analysis. G3: Genes, genomes, genetics, 2016, 6(3): 509-519.
- Nadeau NJ, Martin SH, Kozak KM, et al.Genome-wide patterns of divergence and gene flow across a butterflyradiation. Mol. Ecol., 2013, 22: 814–826.


