What is Bacterial transformation
Bacterial transformation denotes the process of introducing plasmids into bacterial cells to amplify the plasmids or express the genes carried by the plasmids within the bacterial host. Typically, plasmids harboring resistance genes are introduced into competent bacterial cells via chemical or electroporation-based methods. Subsequently, the bacterial culture is spread onto agar plates containing the appropriate antibiotic, thereby selecting for bacteria carrying the desired plasmid.
Bacterial Transformation Principles and Procedural Steps
The fundamental principles and procedural steps involved in bacterial transformation are commonly elucidated through chemical transformation, often referred to as the heat shock method. The focus herein revolves around this approach.
The basic principle underlying bacterial transformation entails the ability of DNA molecules to enter bacterial cells when they are in a state of competence within a CaCl2 solution of low osmolarity. DNA molecules adhere to the cell surface and undergo a brief heat shock treatment at 42°C, which facilitates the absorption of DNA complexes by the cells. Subsequently, the transformed bacteria are cultured in non-selective media for a period to allow the expression of newly acquired phenotypes, such as ampicillin resistance, attained during the transformation process. Following this, the bacterial culture is plated onto selective media containing ampicillin. Finally, colonies that emerge are screened to identify positive clones.
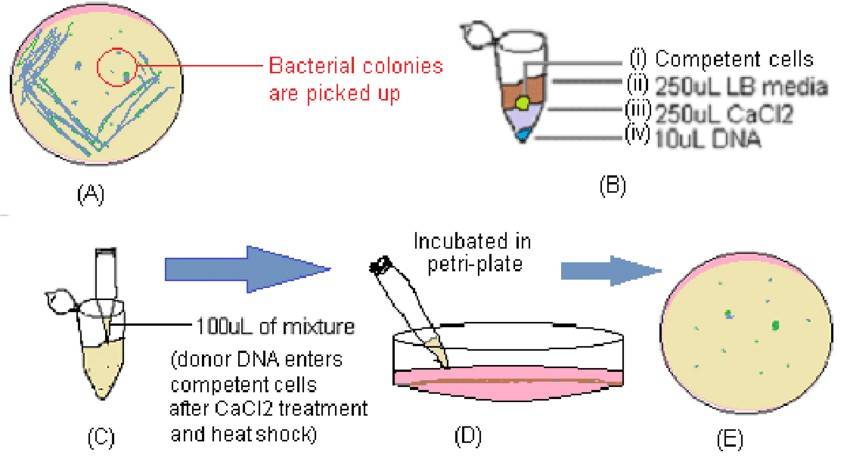 Illustration of main stages of bacterial transformation protocol (Ratul Banerjee 2022)
Illustration of main stages of bacterial transformation protocol (Ratul Banerjee 2022)
Introduction to Competent Cells:
In the realm of molecular biology, the term 'competent cells' alludes to bacterial cells that exhibit an augmented permeability, thereby facilitating the influx of exogenous DNA into the cell's cytoplasm. Under typical conditions, the cell membrane establishes a considerable impediment to the admittance of foreign genetic matter. Nevertheless, the employ of specialized methodologies, including the treatment of cells with calcium chloride (CaCl2), permits modulation of this cellular permeability. Theoretically, CaCl2 treatment incites the formation of ephemeral pores, also referred to as fenestrations, on the cell membrane, thus enabling the ingress of external genetic elements, or vectors, into the competent cells. Worth noting is the fact that due to the dynamic nature of the cell membrane, these fenestrations serve as fleeting passageways, and the membrane retains inherent recuperative mechanisms to reinstate its structural integrity.
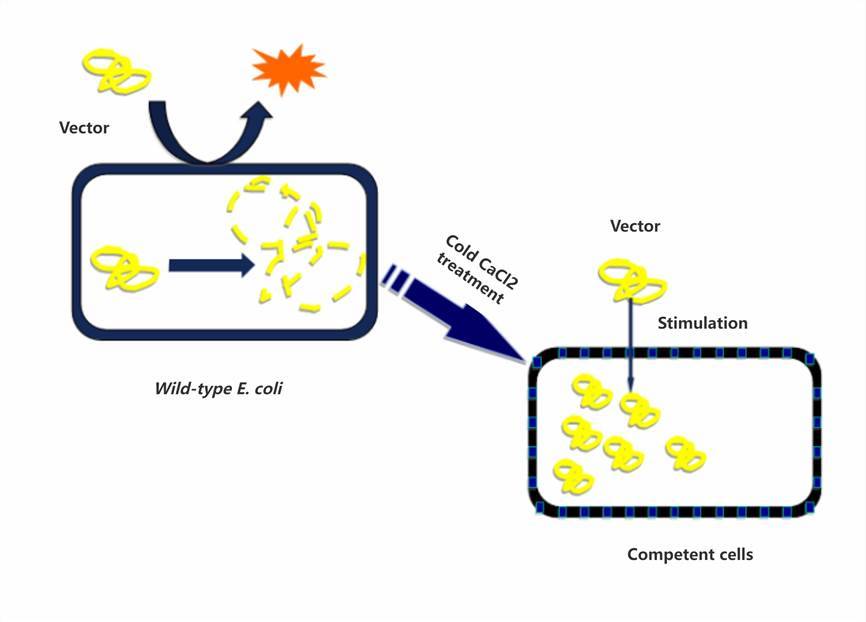
Bacterial Transformation Steps
Preparation of Competent Cells (Part 1): Bacterial Activation and Expansion
How do we obtain competent cells? Specifically, the process begins with the activation of Escherichia coli (E. coli) culture. To achieve this objective, the following steps are undertaken: a small volume of bacterial culture is inoculated into a test tube containing 4 mL of antibiotic-free Luria-Bertani (LB) broth and placed in a shaking incubator at 37°C for overnight incubation, yielding a fresh E. coli culture. Subsequently, the activated culture is inoculated into a centrifuge tube containing 50 mL of antibiotic-free LB broth for scale-up cultivation. Throughout the cultivation process, close monitoring of the culture's status is imperative. Upon the appearance of turbidity in the culture, optical density at 600 nm (OD600) readings should be taken every 20 to 30 minutes. Cultivation should be promptly halted when the OD600 value reaches approximately 0.6.
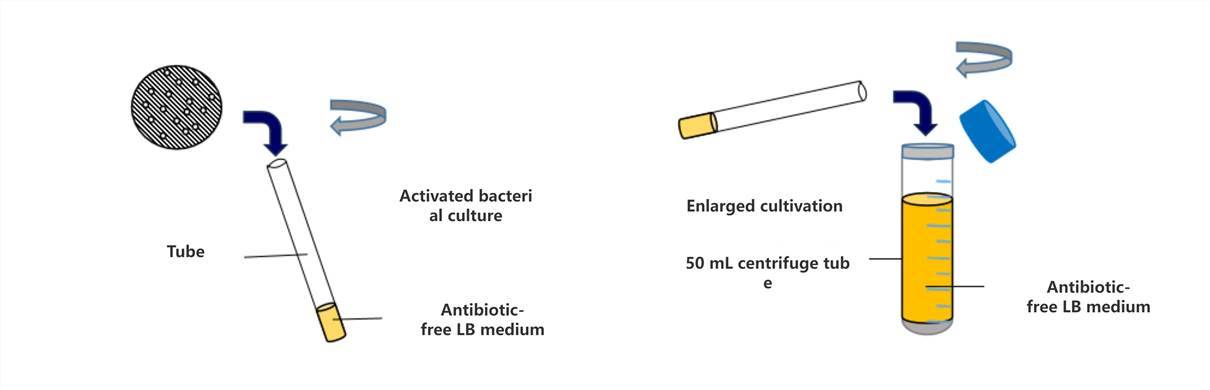
The preserved bacterial strains are resuscitated on antibiotic-free LB agar plates, followed by single colony selection and activation.
Upon completion of activation, scale-up cultivation is performed using large-volume containers such as conical flasks or centrifuge tubes.
Preparation of Competent Cells (Part 2): Preparation
Subsequently, the culture suspension is withdrawn and subjected to a 10-minute ice bath, followed by centrifugation at 5000 revolutions per minute (rpm) for 10 minutes at 4°C. The supernatant is then discarded, and the bacterial cells are gently resuspended in 5 mL of ice-cold CaCl2 solution to ensure uniformity, followed by incubation on ice for 30 minutes. Subsequent centrifugation at 5000 rpm for 10 minutes at 4°C is performed, followed by discarding the supernatant and gentle resuspension of bacterial cells in 5 mL of ice-cold CaCl2 solution to achieve homogeneity. After brief incubation on ice, the competent cell suspension is prepared. It is aliquoted into 1.5 mL centrifuge tubes, preferably utilized promptly for transformation, or alternatively stored for future use under ultra-low temperature conditions at -70°C.
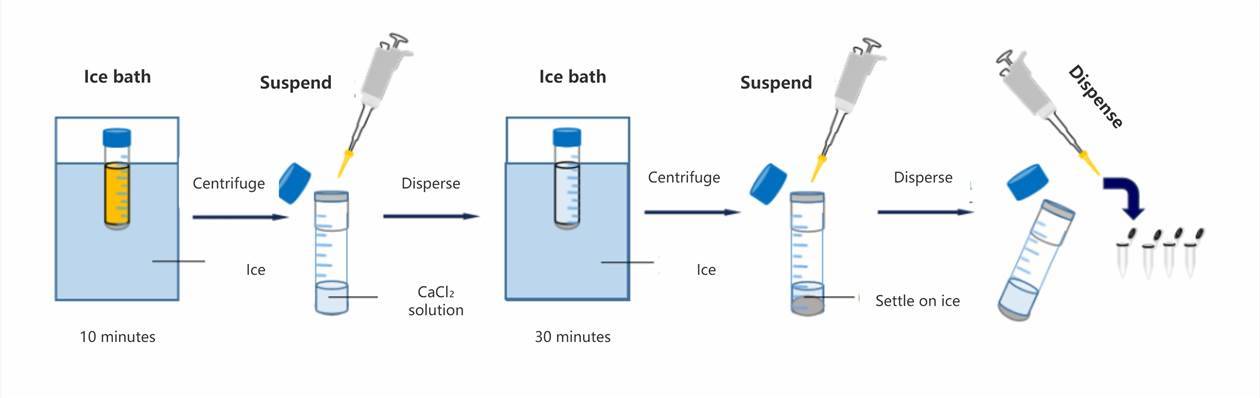
Pre-cool instruments such as centrifuges and reagents like CaCl2 solution before commencing experiments.
Perform all procedures swiftly on ice to prevent warming and contamination.
Preparation of Recombinant Vectors
Subsequently, we delve into the intricacies of the heat shock transformation process. In the standard protocol of bacterial transformation, the construction of vectors stands as the pivotal objective, necessitating the efficient fusion of target genes with the vector. Typically, we opt for restriction enzymes specific to multiple cloning sites on the vector, enabling dual enzyme digestion of both the vector and the target gene fragments. Subsequently, employing T4 ligase, the digested vector and target gene fragments are precisely ligated to attain the desired fusion product. This process demands meticulous manipulation and stringent control to ensure both efficiency and accuracy of the ligation process.
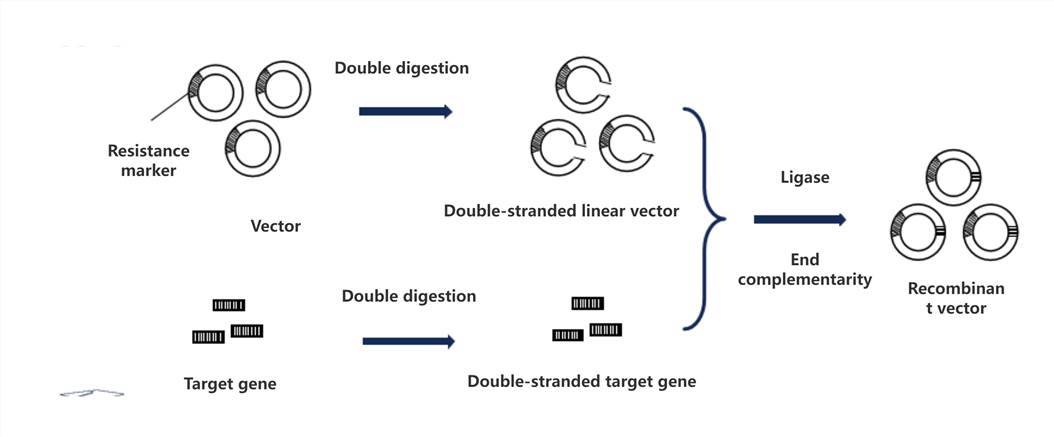
Fragments of the vector are simultaneously cleaved! Cohesive endonucleases incise both the vector and target gene (or compatible ends enzymes).
Complementary ends unite! Ligases facilitate the dual-end complementary joining of the vector and target gene.
Thermal Induction of Competent Cell Transformation
The transformation mixture, containing the carefully prepared recombinant product, is gently introduced into competent cells, ensuring even distribution. Subsequently, the mixture is incubated on ice for 30 minutes to facilitate cell adhesion to the recombinant product. Following this, the mixture is swiftly transferred to a preheated water bath set to 42°C for a 90-second heat shock, inducing cellular uptake of the recombinant product. Immediately post-heat shock, the mixture is returned to ice and allowed to cool for 2 minutes to prevent thermal damage. Subsequently, 700 microliters of antibiotic-free LB liquid culture medium is added, and the mixture is gently mixed. Finally, the mixture is incubated in a shaking incubator at 37°C for 40 minutes to allow the recipient cells to resume normal growth and facilitate the generation of transformants with the desired resistance characteristics.
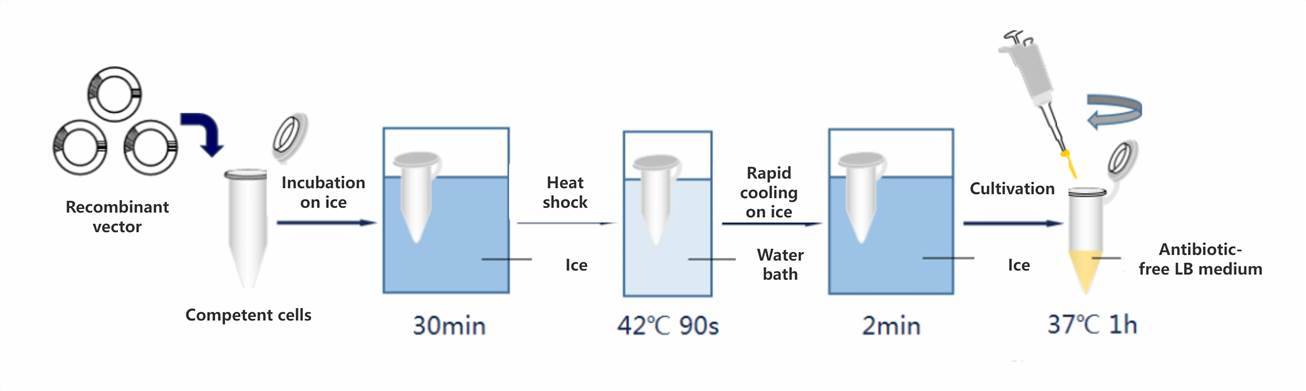
The alternation between cold and heat stimuli induces changes in the permeability of sensory cells.
Upon exposure to heat stimulation, sensory cells become activated, while they persist within the cellular body following the restoration of permeability during cold immersion.
Application of Recombinant Culture Suspension
The cultured bacterial suspension, following amplification, is subjected to centrifugation at a continuous rate of 3000 revolutions per minute for 5 minutes. Subsequently, the majority of the supernatant is removed, leaving approximately 100 microliters for resuspension. This resuspended volume is uniformly spread onto the surface of solid LB agar containing specific antibiotics. Finally, the prepared agar plates are placed in a constant temperature incubator at 37°C and allowed to incubate undisturbed for 12 to 16 hours.
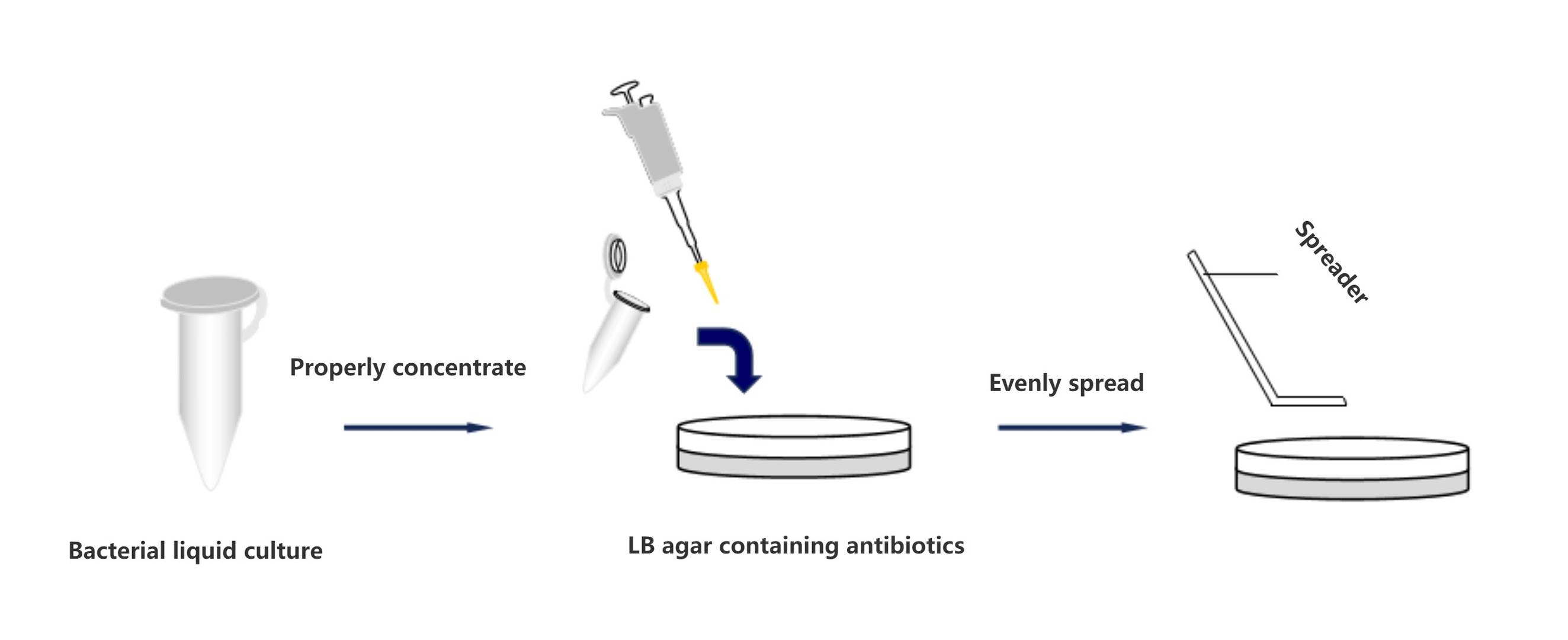
The incubation period for LB medium without antibiotics should be carefully regulated to prevent overgrowth of Gram-negative bacterial strains.
Uniform spreading and appropriate volume application are essential during plating to ensure the formation of an adequate number of individual bacterial colonies.
Screening of Recombinant Colonies
Upon formation of bacterial colonies, validation of their positivity is typically conducted using colony PCR methodology.
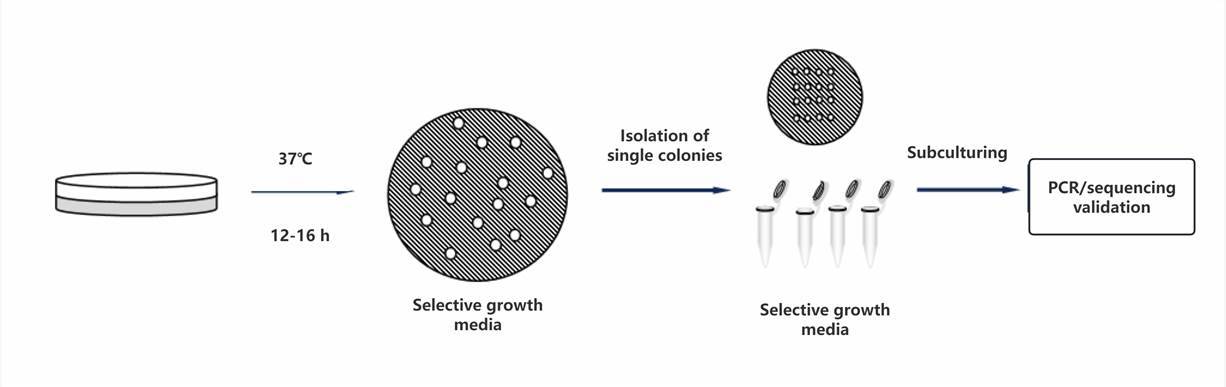
Different bacterial strains require varying incubation temperatures and durations to facilitate the formation of individual colonies as a guiding principle.
Positive recombinant colonies are preserved; therefore, it is customary to first conduct single colony expansion.
Analysis of Bacterial Transformation Results
Verification of Recombinant Colonies via PCR
The objective of bacterial transformation is to obtain recombinant vectors harboring the target gene, ultimately validated through PCR. As illustrated in the figure, bands of 750 bp in lanes 1, 3, 5, 6, 7, and 8 correspond to the desired gene size, indicating positive clones, whereas lanes 2 and 4 represent false positives. Positive clones are further subjected to sequencing to confirm the composition of the recombinant plasmid.
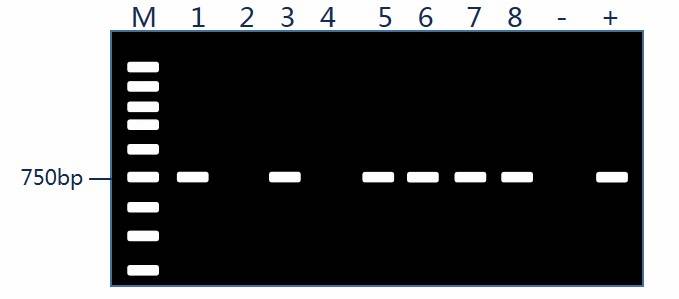
While PCR is a rapid and effective method, it has certain limitations. Firstly, the positivity confirmed by PCR does not guarantee the accuracy of the recombinant construct; however, failure to verify by PCR can generally be construed as negativity. To minimize false positives, it is advisable to design primers targeting the junction between the vector and the insert during PCR validation.
Furthermore, PCR can only detect known sequences. Therefore, if you are solely concerned with the presence of specific genes or sequences, PCR might suffice. However, if you seek to confirm the integrity of the entire exogenous DNA fragment, the accuracy of the sequence, and the detection of unknown mutations or variations, PCR may not provide sufficient information.
At this juncture, sequencing becomes paramount. Through sequencing, one can acquire the sequence information of the entire exogenous DNA fragment, ensuring its integrity and accuracy. Additionally, sequencing can unveil potential unknown mutations or variations, furnishing a more comprehensive and detailed dataset that aids in validating the accuracy and reliability of the transformation process.
Bacterial Transformation Verification via Sequencing
The Importance of DNA Sequencing in Genetic Transformation Experiments
Genetic transformation, the process of introducing foreign DNA into an organism, stands as a cornerstone technique in molecular biology. Yet, the imperative of DNA sequencing post-transformation hinges upon several determinants. Let us delve into the considerations steering this decision-making process.
Objective of the Experiment: When aiming to introduce specific mutations, inserts, or construct novel plasmids, sequencing emerges as an imperative. This pivotal step verifies the presence and accuracy of the intended genetic modifications.
Reliability of Cloning Method: Varied cloning techniques exhibit differing error rates. While high-fidelity methods may mitigate errors, sequencing remains prudent, particularly for intricate constructs or when mutations are introduced.
Previous Validation of Constructs: Constructs with well-documented reliability under analogous conditions may obviate the need for sequencing after each transformation. Routine procedures boasting consistent performance may bypass this stage.
Screening Strategies: High-throughput screening strategies may render sequencing every clone impractical. Initial screening methods such as functional assays or restriction analysis can winnow down candidates, with sequencing reserved for chosen clones to affirm sequence fidelity.
Resource Considerations: Sequencing entails time and cost. The decision to sequence should factor in resource availability and the experiment's criticality. Crucial or innovative experiments may warrant sequencing investment to ensure dependable outcomes.
Regulatory and Publication Requirements: Experiments earmarked for regulatory submission or publication typically mandate sequencing to meet exacting standards. Confirming the identity and integrity of transformed DNA stands pivotal for scientific reporting.
In summation, though not obligatory for every transformation, DNA sequencing constitutes a pivotal stride in affirming genetic alterations and ensuring experimental robustness. The decision to sequence should carefully balance experimental objectives, cloning method reliability, resource availability, and downstream applications of the transformed material.
Leveraging Whole Genome Sequencing and Plasmid Sequencing in Bacterial Transformation Studies
Whole Genome Sequencing (WGS): WGS delivers an all-encompassing perspective on an organism's complete genetic constitution, inclusive of chromosomal DNA and extrachromosomal components such as plasmids. In the realm of plasmid transformation, WGS is instrumental for several indispensable purposes:
Identification of Plasmid Integration: WGS facilitates the accurate detection of exogenous plasmid integration sites within the bacterial genome. By contrasting the sequenced genome of the transformed cells with that of the unmodified, wild-type strain, investigators can accurately locate the specific loci of plasmid insertion.
Examination of Genetic Modifications: Through WGS, genetic modifications instigated by plasmid transformation - encompassing single nucleotide polymorphisms (SNPs), insertions, deletions, or genomic rearrangements – can be portrayed. This vital information assists in assessing the global impact of transformation upon the host genome.
Determination of Transformation Effectiveness: Quantitative dissection of sequencing data endows researchers with the capacity to ascertain the extent of successful plasmid integration events. This reveals insightful information concerning transformation efficiency and surrounding experimental condition variability.
Plasmid Sequencing: Plasmid Sequencing is an investigative procedure specifically designed to extrapolate the sequence and architecture of plasmid DNA. Incorporated into plasmid transformation studies, it confers several benefits:
Validation of Plasmid Integrity: Plasmid Sequencing certifies the integrity and accuracy of the plasmid vector both prior to and following transformation, thus verifying its preservation against unexpected mutations or deletions arising during the cloning or propagation stages.
Recognition of Insertion Sites: Plasmid Sequencing allocates in-depth information related to the sequences on either side of the cloning sites within plasmids. This data serves as a reliable source for verifying if foreign DNA inserts at the desired regions within the bacterial genome.
Characterization of Plasmid Variations: Plasmid Sequencing supports the identification and characterization of plasmid variations, consisting of modified replication origins, antibiotic resistance markers, or distinct genetic elements. Comprehension of plasmid diversity enriches researchers' potentiality to devise specialized vectors for appointed applications.
Conclusion: The incorporation of Whole Genome Sequencing and Plasmid Sequencing within plasmid transformation studies ensures a thorough comprehension of genetic modifications, integration events, and transformation effectiveness. These sophisticated sequencing methodologies not merely endorse the successful delivery of plasmid, but also shed light on the optimisation of transformation protocols and the customization of unique genetic constructs. Engaged as revolutionary tools within molecular biology, WGS and Plasmid Sequencing provide the momentum for innovative progression within the sphere of genetic engineering.
Analysis of Difficulties in Bacterial Transformation
Following bacterial transformation, the growth pattern of colonies on agar plates sometimes deviates from the norm, presenting several potential scenarios:
The first scenario entails the absence of colonies on the plate. This occurrence may stem from various factors, including DNA degradation, inactivation of competent cells, plasmid resistance interference, or bacterial death due to inadequate cooling of the glass rod used during streaking. Each potential cause necessitates meticulous investigation.
The second scenario involves an excessive number of colonies, rendering isolation of single clones unfeasible. This situation may arise from overly concentrated bacterial suspensions, warranting the application of a minimal volume during plating.
Uneven colony distribution constitutes the third scenario, primarily attributable to uneven plating techniques.
The presence of contaminants on the plate constitutes the fourth scenario, potentially arising from expired or ineffective antibiotics or contamination of competent cells.
The fifth scenario manifests as low transformation efficiency, evidenced by sparse colony growth. This occurrence may result from inadequate amounts of transforming DNA, low competence efficiency, or failure to sufficiently cool the glass rod during plating.
References
- van Belkum A, Dunne WM Jr. Next-generation antimicrobial susceptibility testing. J Clin Microbiol. 2013
- Dugar G, Herbig A, Förstner KU, Heidrich N, Reinhardt R, Nieselt K, Sharma CM. High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates. PLoS Genet. 2013
- Oh JH, van Pijkeren JP. CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res. 2014
- omoiaga, D., Bubnell, J., Herndon, L. et al. High rates of plasmid cotransformation in E. coli overturn the clonality myth and reveal colony development. Sci Rep 12, 11515 (2022).