
In recent years, the problem of antibiotic resistance has developed into a global public health crisis. According to the data of the World Health Organization (WHO), the number of deaths caused by antibiotic resistance continues to rise every year. If effective intervention measures are not taken, it is estimated that by 2050, more than 10 million people around the world will lose their lives every year. As the genetic basis for the spread of drug resistance, antibiotic resistance genes (ARGs) can spread rapidly among different microorganisms through horizontal gene transfer, widely exist in various environmental media such as soil, water, hospital sewage, etc., and even form a drug resistance gene pool in the human intestinal flora, which aggravates the complexity and prevention and control difficulty of drug resistance risk.
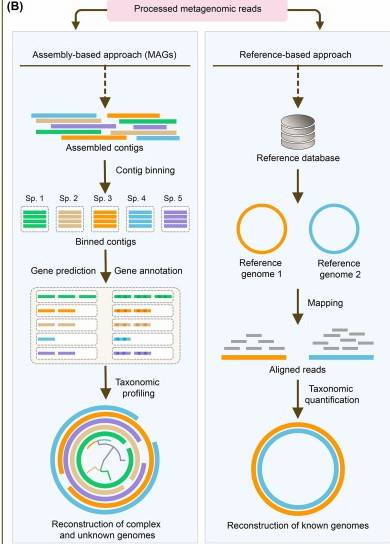
Although the detection technology for ARGs has evolved from traditional culture methods to molecular biology methods, such as metagenome sequencing, existing research still faces multiple challenges. The diversity and abundance of ARGs in environmental samples differ significantly, and various detection methods have limitations in sensitivity, specificity, and flux, which makes it difficult to achieve accurate traceability and dynamic monitoring of ARGs. In addition, the synergistic mechanism of ARGs with host bacteria and environmental factors is not completely clear, which greatly limits the formulation of effective prevention and control strategies. Therefore, it is of great practical significance to develop a systematic and efficient ARG analysis solution to reveal the law of drug resistance spread and block the ARG diffusion path.
Through two case studies, this paper analyzes the transmission mechanism, global diversity and driving factors of ARGs in sewage treatment system, and looks forward to the future development direction of ARGs analysis.
Exploration on the Spreading Mechanism of ARGs
Title: Genetic compatibility and ecological connectivity drive the dissemination of antibiotic resistance genes
Publish Magazine: Nat Commun
Impact Factors: 15.7
Publication Time: 2025.03.16
DOI: https://doi.org/10.1038/s41467-025-57825-3
The spread of ARGs through horizontal gene transfer has become a major threat to global public health. However, the transmission mechanism of ARGs between pathogens is still poorly understood. Recently, researchers identified ARGs-level gene transfer events in about 1 million bacterial genomes through phylogenetic analysis, and combined with more than 20,000 metagenome data from animal, human, soil, water, and wastewater microbiota, constructed a random forest model that can predict ARGs-level gene transfer among bacteria.
Studies have shown that genetic incompatibility (difference in nucleotide composition) will reduce the transfer probability of ARGs between evolutionarily divergent strains, while environmental co-occurrence (especially in the human body and wastewater environment) will significantly increase the transfer possibility and present a specific environmental transmission mode. This study establishes a data-driven prediction method of ARG propagation and provides new insight for analyzing the regulation mechanism of this evolutionary process.
 Overview of the analysis pipeline (Lund et al., 2025)
Overview of the analysis pipeline (Lund et al., 2025)
Identification of Resistance Genes to Horizontal Metastasis
Through the systematic analysis of 867,318 bacterial genomes, more than 2.66 million ARGs involving 10 types of resistance mechanisms were identified, mainly aminoglycosides (40.9%) and β -lactams (38.4%). Based on phylogenetic analysis, 6,276 cases of horizontal metastasis were identified, among which aminoglycoside phosphotransferase (APHs) (29.9%) and A/C/D β-lactamase (23.8%) were the most common. It is worth noting that there are significant differences in the similarity of transferred genes: some genes (APHs, Erm 23S rRNA methyltransferase, tetracycline efflux pump, etc.) remain highly conserved (> 99% amino acid consistency), while some genes (aminoglycoside acetyltransferase (AACs), A/C/D β -lactamase, etc.) show great sequence differences.
 Summary of identified horizontal transfers of ARGs, and performance of random forest models trained to predict horizontal ARG transfer (Lund et al., 2025)
Summary of identified horizontal transfers of ARGs, and performance of random forest models trained to predict horizontal ARG transfer (Lund et al., 2025)
Predicting the Horizontal Transfer of ARGs
A machine learning model based on random forest was established, and the horizontal gene transfer of ARGs between bacterial hosts was predicted through genetic incompatibility (genome 5-mer difference, gene-genome 5-mer difference, and host genome size difference), environmental co-occurrence (five environments of animals, human body, soil, water and wastewater), host's Gram staining characteristics and the characteristics of transferred ARGs genes.
It was found that the difference in nucleotide composition between genomes and between genomes and -ARGs had a significant negative impact on the possibility of gene transfer (especially tetracycline efflux pump and ribosome protection protein coding gene (RPGs)), and the difference of Gram staining characteristics also inhibited gene transfer (except that erm gene and tetracycline RPGs were easier to transfer among Gram-positive bacteria). On the contrary, environmental co-occurrence significantly promotes gene transfer, with the most significant impact on human microbiota (transfer of aminoglycoside phosphotransferase (APHs) and A/C/D β-lactamase), followed by wastewater environment (transfer of AACs).
 Relative importance of genetic and environmental factors for predicting the horizontal transfer of antibiotic resistance genes (Lund et al., 2025)
Relative importance of genetic and environmental factors for predicting the horizontal transfer of antibiotic resistance genes (Lund et al., 2025)
Genetic Incompatibility Inhibits the Transfer of ARGs
By constructing a partial dependence graph, it was found that the differences in nucleotide composition (Euclidean distance of 5-mer distribution) between host genomes and between ARGs- genomes were negatively correlated with the horizontal transfer probability, and the initial thresholds of the negative effects were roughly corresponding to the differences in the median nucleotide composition among hosts of different bacterial classes and the typical differences in the composition of ARGs and different phylum host genomes. Among them, the transfer of tetracycline efflux pump coding genes is extremely sensitive to gene-genome incompatibility.
It is worth noting that although different phylum hosts usually present high genetic differences, the nucleotide similarity of some cross-phylum host combinations (such as Bacillota and Bacteriodota) is higher than that of some intraphylum hosts, indicating that high genetic incompatibility may not completely block the horizontal transfer of ARGs from distant hosts, and its spread may be restricted by other factors (such as the host range carrying the movable genetic elements of ARGs).
 Relative contribution of genetic incompatibility for prediction of the horizontaltransfer of antibiotic resistance genes (Lund et al., 2025)
Relative contribution of genetic incompatibility for prediction of the horizontaltransfer of antibiotic resistance genes (Lund et al., 2025)
Promoting ARGs Transfer in Human and Wastewater
By constructing the co-occurrence network of host bacteria, it was found that 63.3% of high-frequency gene transfer host pairs (≥5 transfers) co-existed in at least one kind of environment. The human microbiome shows the richest co-occurrence host diversity, Including Pseudomonas (such as Escherichia coli and Acinetobacter baumannii), There are many kinds of pathogens in the phylum Bacillota (such as Staphylococcus aureus) and Campylobacterota (such as Campylobacter jejuni), in which the intestinal tract and skin sub-environment can promote the transfer of macrolides, tetracyclines, aminoglycosides and β -lactams ARGs respectively.
In contrast, although the co-occurrence ratio of wastewater microbiota hosts is higher, the classification range is more limited, mainly involving Gammaproteobacteria (such as Acinetobacter and Pseudomonas) and some strains of Firmicutes. It is worth noting that 23.8% of the metastasis-related hosts co-occurring in the human microbiota were not detected in the wastewater microbiota, whereas the proportion was only 3.3%.
Studies have shown that 12 species of ARGs, such as Klebsiella pneumoniae and Acinetobacter baumannii, have frequently participated in horizontal gene transfer, and the co-occurrence of these bacteria with their common transfer partners is the most intensive in human microbiota and wastewater, which is much higher than that in animal, soil and water environment.
 Co-occurrence of promiscuous bacterial taxa in human and wastewater microbiomes (Lund et al., 2025)
Co-occurrence of promiscuous bacterial taxa in human and wastewater microbiomes (Lund et al., 2025)
Investigating the Mobility of ARGs in Human Sewage Treatment System
Title: Global diversity and distribution of antibiotic resistance genes in human wastewater treatment systems
Publish Magazine: Nat Commun
Impact Factors: 15.7
Publication Time: 2025.04.29
DOI: https://doi.org/10.1038/s41467-025-59019-3
About 52% of the world's population sends sewage to sewage treatment plants, which is an important infrastructure to protect human and ecosystem health. However, sewage treatment plants are one of the most important repositories of ARGs and ARBs, because they receive wastewater from homes, hospitals, and pharmaceutical facilities. Most sewage treatment plants adopt an activated sludge process (AS), that is, an open aerobic enrichment culture system for microbial flocs or particles.
Few studies have evaluated the environmental factors that drive resistance in sewage treatment plants. Therefore, the global diversity of ARGs in sewage treatment plants and the understanding of the potential mechanism affecting ARGs in sewage treatment plants are still incomplete. In order to understand the global situation of ARGs in sewage treatment plants, it is necessary to conduct a systematic, consistent, and globally representative investigation.
 The abundance and global distribution of the AS resistomes (Zhu et al., 2025)
The abundance and global distribution of the AS resistomes (Zhu et al., 2025)
Relationship Between AS Resistance Group and Bacterial Community
In order to understand the relationship between the resistance group and bacterial community structure, we conducted a Procrustes analysis. The bacterial community structure is represented by a 16S rRNA gene extracted from a metagenome or amplified 16S rRNA gene. According to Procrustes analysis, the matrix-matrix correlation coefficient of bacterial community structure based on metagenome 16S is 0.74, and the matrix-matrix correlation coefficient of bacterial community structure based on 16S amplicon is 0.70, which indicates that there is a strong correlation between bacterial community structure and resistance groups in sewage treatment plants.
In order to further determine whether the relationship between the resistant group and the microbial group depends on the phylogenetic tree, we determined the relationship between the total ARG abundance and the relative abundance of the first four major ARG groups and the main phylum. Bacteroides is the most abundant phylum, which is positively correlated with the ARG abundance based on the data of the 16S rRNA gene in amplicon. Based on the 16S rRNA gene from the metagenome, the abundance of ARGs is also positively correlated with Chlorobacter, Acidophilus, Blastomycetes, Nitrospira, and δ Proteobacteria, indicating that these groups may be the main carriers of ARGs. These results indicate that the resistance group in AS may be closely related to microbial physiology.
 The linkage of the AS resistomes to microbiomes (Zhu et al., 2025)
The linkage of the AS resistomes to microbiomes (Zhu et al., 2025)
Resistance Group and Mobility of MAG
MGEs promoted the horizontal transfer of ARGs and contributed to the spread and evolution of antibiotic resistance in microbial communities. In order to determine the diversity of MGE, a total of 2200 non-redundant ORFs were identified as 56 MGE genes. The three MGEs with the highest abundance in AS are tnpA, IS91, and tniA respectively. The total MGE abundance shows significant differences among six continents and different countries. Linear regression showed that MGE richness was positively correlated with ARG richness. In addition, the total abundance of ARG is positively correlated with the abundance of nearby MGEs.
Furthermore, the mobility is quantified according to the ARGs shared between different hosts. The mobile ARGs were identified as identical or nearly identical sequences in different bacterial hosts. From these 112 replicative MAGs, 3646 ORFs were annotated as ARG sequences, and they were further clustered into 2368 ARG clusters with 99% nucleotide consistency. Subsequently, 29% (682/2368) of 54% ARG clusters covering all ARG sequences (1959/3646) were distributed to multiple species, which indicated that there may be horizontal gene transfer across distant organisms recently. In contrast, 10% of the ARG clusters in MAGs are multi-species ARGs. The proportion of potentially mobile ARGs in AS is significantly higher than that in human microbiota.
 Mobility of ARGs from assembly and MAG-based analyzes (Zhu et al., 2025)
Mobility of ARGs from assembly and MAG-based analyzes (Zhu et al., 2025)
Driving Factors of Global AS Resistance Group
We used a standardized random rate (NST) to quantitatively evaluate the relative contribution of stochastic process and deterministic process to the global AS-resistant genome changes. Except in Europe, the estimated NST of drug-resistant genomes in all continents is generally above 0.5, indicating that stochastic processes may play a role in the changes in AS drug-resistant genomes. The analysis of variance decomposition (VPA) based on matrix multiple regression also showed that 67.4% of the resistance group variation could not be explained by the measured environmental variables and geographical distance. These results are consistent with previous findings, that is, stochastic processes are important in shaping bacterial community assembly.
In order to further identify the role of a single deterministic factor, we use a univariate model to test the environmental variables that have a significant correlation with the change in ARG abundance. Mixed liquor-suspended solids (MLSS), temperature, and urban population are positively correlated with ARG abundance. On the contrary, the abundance of ARG is negatively correlated with pH, solid residence time, and influent biochemical oxygen demand (BOD), which are reported to play an important role in regulating the community structure of AS bacteria. Different from the previous observation that there is a strong correlation between the abundance of ARGs in sewage and socio-economic factors, we find that there is no significant correlation between the abundance of ARGs in the sewage treatment plant and the per capita GDP and the number of antibiotics used in the country. This non-correlation may indicate that the concentration of antibiotics in AS may not be enough to exert significant selective pressure on the maintenance and spread of ARGs.
 Drivers for the AS resistomes (Zhu et al., 2025)
Drivers for the AS resistomes (Zhu et al., 2025)
Conclusion
These cases not only show the great potential of collaborative application of microbiology, metagenome sequencing technology, and bioinformatics tools but also provide a replicable model for global public health and environmental governance. Although there are still challenges in data standardization, integration of cross-regional monitoring systems, and analysis of new drug resistance mechanisms, the innovative model of multi-party linkage between doctors and doctors in Industry-University-Research is pushing the ARG research from theoretical exploration to precise prevention and control practice.
With the deep integration of nanotechnology, artificial intelligence algorithm, and synthetic biology, ARG analysis will be iteratively upgraded in the direction of high sensitivity, Qualcomm, and real-time dynamic monitoring in the future, to build a scientific and technological defense line to break the chain of drug resistance and protect human health and ecological security.
References
- Lund D, Parras-Moltó M, Inda-Díaz JS, et al. "Genetic compatibility and ecological connectivity drive the dissemination of antibiotic resistance genes." Nat Commun. 2025 16(1): 2595 https://doi.org/10.1038/s41467-025-57825-3
- Zhu C, Wu L, Ning D, et al. "Global diversity and distribution of antibiotic resistance genes in human wastewater treatment systems." Nat Commun. 2025 16(1): 4006 https://doi.org/10.1038/s41467-025-59019-3





 A"
width="400" height="200" loading="lazy"
alt="Antibiotic Resistance Genes (ARGs): Detection, Database, and Bioinformatics Tools">
A"
width="400" height="200" loading="lazy"
alt="Antibiotic Resistance Genes (ARGs): Detection, Database, and Bioinformatics Tools">


