Identifying Antimicrobial Resistance and Virulence Factors Using Next-Generation Sequencing
Inquiry >Overview of NGS
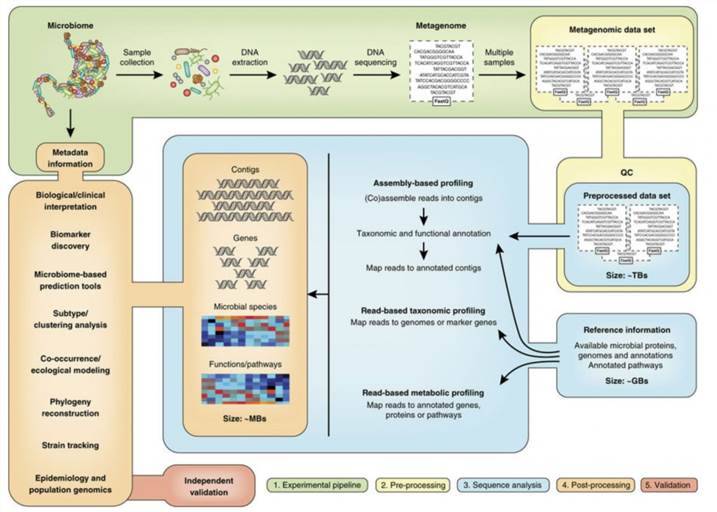
Scientists have attempted to advance sequencing technology since the sequencing of the first bacterial virus by Sanger et al. in 1977, and have created new methods and sequencers that enable high throughput sequencing. More than 60,000 sequencing projects have been made available to the public since then. These include more than 49,000 projects on bacterial genomes, 61.2% of which are accessible to public libraries. Thanks to the numerous currently accessible next-generation sequencing (NGS) technologies and platforms, genome sequencing has a big effect on clinical microbiology by facilitating the development of different sequence-based instruments, in particular molecular detection, serological and genotyping assays. Furthermore, NGS also presents a better way to decode the virulence potential and forecast the antibiotic resistance trend of clinical isolates by having access to the full gene repertoire of a strain.
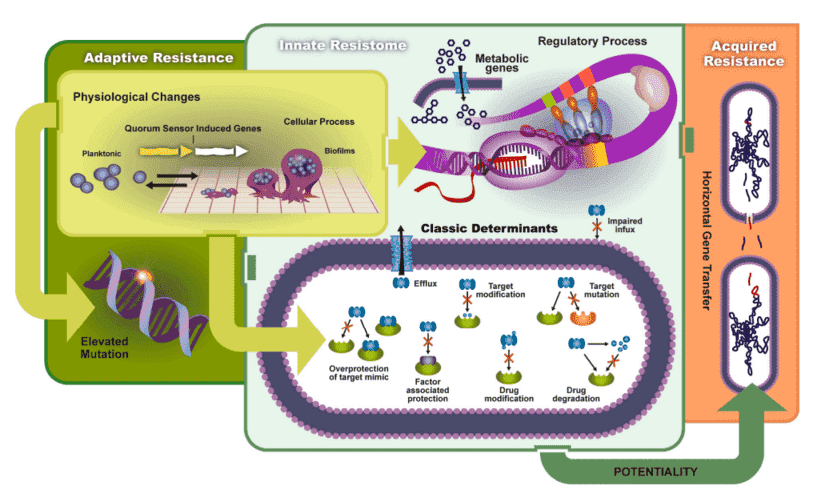 Figure 1. The transmission and mechanisms of antibiotic resistance and virulence can be divided into adaptive resistance, innate resistance, and acquired resistance. (Schroeder, 2017)
Figure 1. The transmission and mechanisms of antibiotic resistance and virulence can be divided into adaptive resistance, innate resistance, and acquired resistance. (Schroeder, 2017)
In studying bacterial pathogenesis and their encounters with the host, and in the development of new medicines, vaccinations, and molecular diagnostic methods, the detection and characterization of virulence factors, especially toxins, and antibiotic resistance markers of pathogens are essential. In addition, it can help to enhance outbreak control and therapeutic intervention by identifying certain virulence or resistance markers.
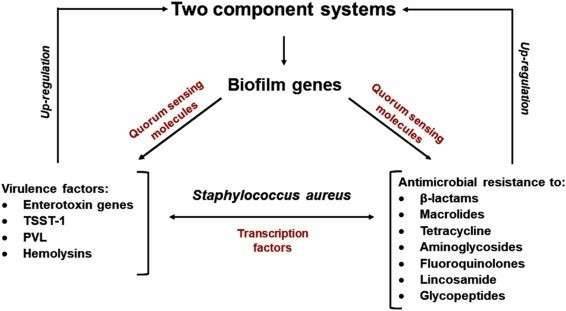 Figure 2. The relationship between virulence factors and antimicrobial resistance in Staphylococcus aureus. (Pérez, 2020)
Figure 2. The relationship between virulence factors and antimicrobial resistance in Staphylococcus aureus. (Pérez, 2020)
Antibiotic Resistance Marker Identification
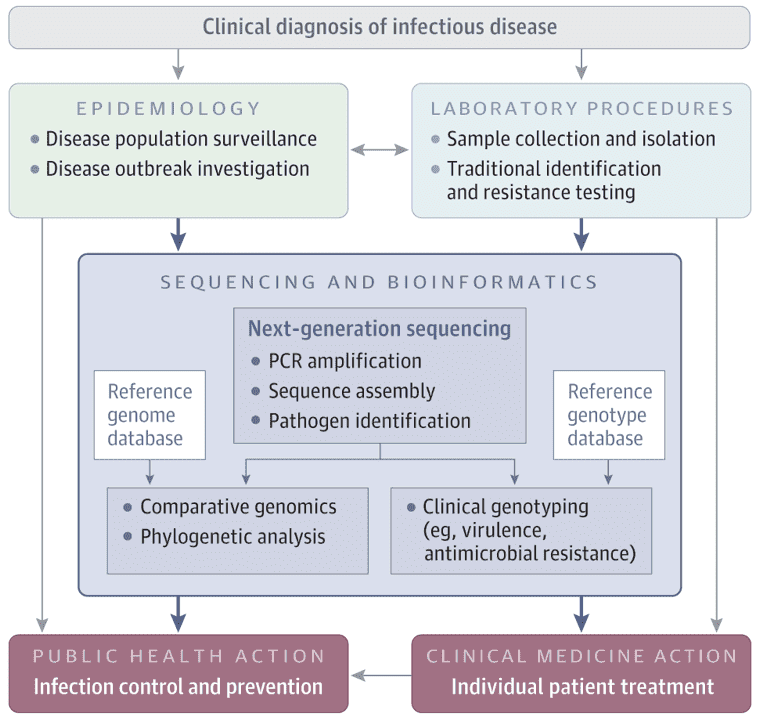
The proliferation of multidrug-resistant (MDR) bacteria has been a global public health issue worldwide over the past few years. Any pathogenic MDR bacteria, including last-resort antibiotics such as imipenem and colistin, are currently immune to all antibiotics used in therapy. Numerous pathogenic MDR bacteria, especially methicillin-resistant S. aureus (MRSA), Enterobacteriaceae, Acinetobacter, and pseudomonas species cause bacterial infections. In order to fully understand resistance pathways, foresee the resistance phenotype, allow effective infection control, improve antibiotic therapy and patient care, design PCR assays to diagnose resistance inducing genes or mutations, recognize targets for novel drugs and before using bacteria that can be used as probiotics, deciphering the resistome of a bacterium has become necessary.
Several mechanisms by which bacteria become immune to antibiotics have been described: development of natural or acquired enzymes that metabolize the antibiotic; production of enzymes that alter antibiotics; alterations of the antibiotic target that avoid its binding; impermeability of the membrane and overexpression of efflux systems. In general, the acquired resistance mechanisms are distributed by MGEs, especially plasmids, transposons, and integrons, and can be identified by genome sequencing.
Virulence Factors Identification
Detection of virulence factors successively depended on biochemical methods prior to genome sequencing, or systemic molecular sampling of a panel of genes revealed that using molecular cloning and/or mutagenesis played a role in pathogenesis. Bacterial toxins or other virulence variables are extracted in biochemical methods and then their pathogenic effects are tested in vivo or in vitro. Virulence genes are studied through mutagenesis and/or cloning and expression of nonpathogenic, often E. coli strains, in molecular approaches. The frequency of virulence factor identification has increased significantly over the past two decades, thanks to genomics along with functional analysis (transcriptomics and proteomics). It is possible to classify bacterial virulence factors in genomes by homology searches for known virulence genes, by matching strains with different degrees of virulence, or by analyzing horizontally acquired genes.
References
- Pérez VK, da Costa GM, Guimarães AS, et al. Relationship between virulence factors and antimicrobial resistance in Staphylococcus aureus from bovine mastitis. Journal of Global Antimicrobial Resistance. 2020.
- Hendriksen RS, Bortolaia V, Tate H, et al. Using genomics to track global antimicrobial resistance. Frontiers in public health. 2019, 7.
- Schroeder M, Brooks BD, Brooks AE. The complex relationship between virulence and antibiotic resistance. Genes. 2017, 8(1).
- Bakour S, Sankar SA, Rathored J, et al. Identification of virulence factors and antibiotic resistance markers using bacterial genomics. Future microbiology. 2016, 11(3).